Patricia Barberá, Robert J. Soreng, Paul M. Peterson, Joan Garcia‐Porta, Konstantin Romaschenko, Carlos Aedo, and Alejandro Quintanar
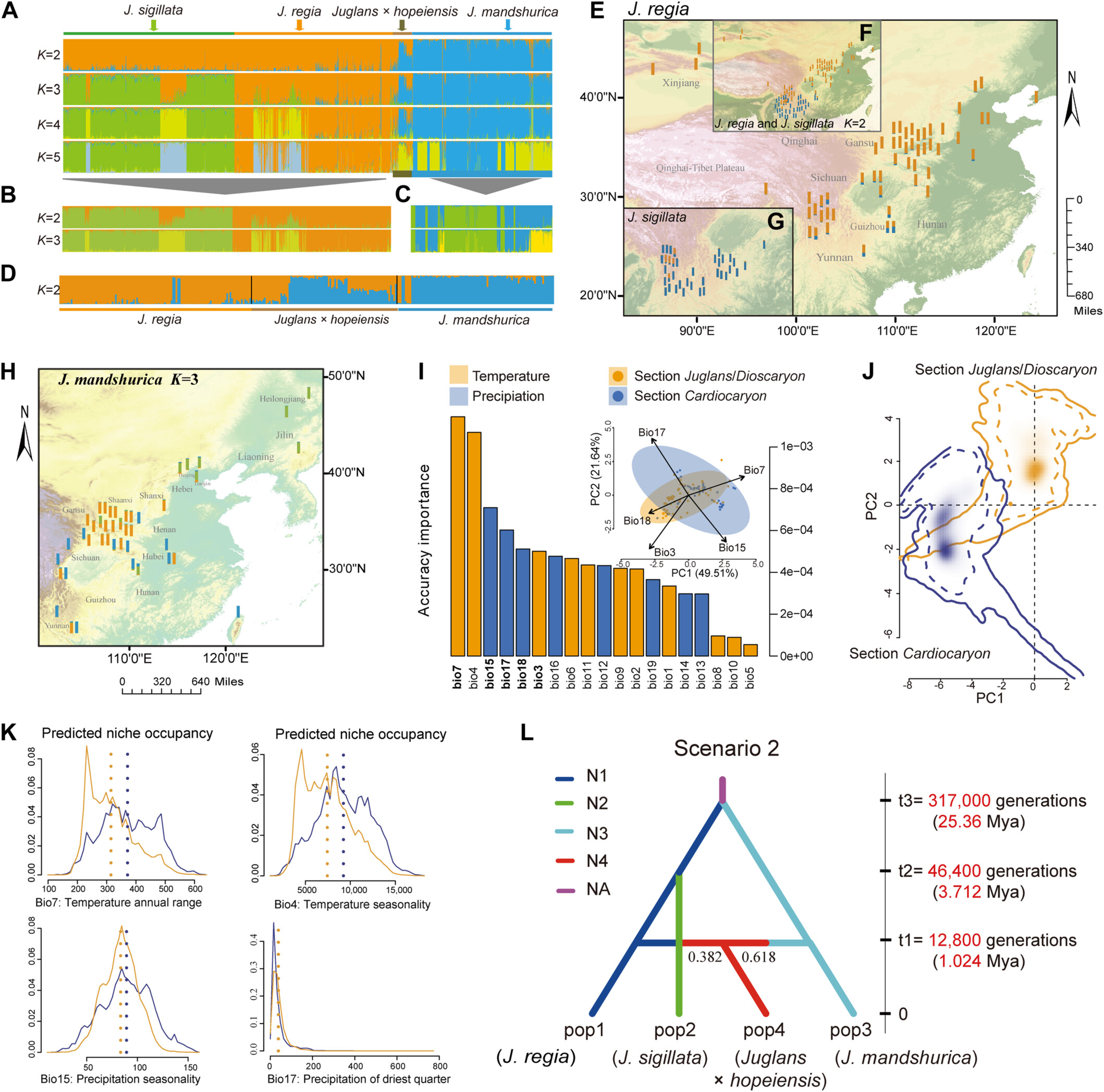
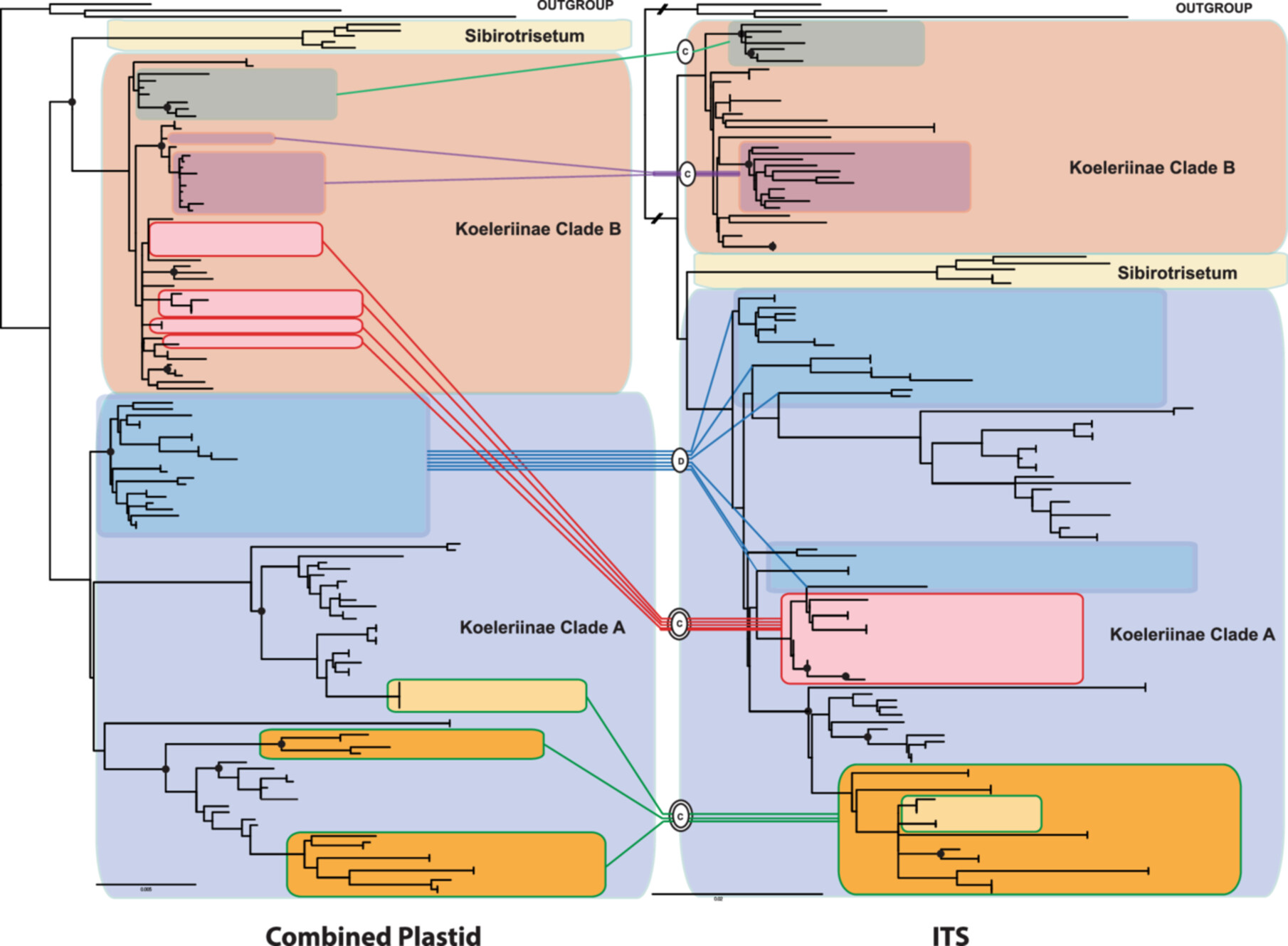
Koelerioid grasses (subtribe Aveninae, tribe Poeae; Pooideae) resolve into two major clades, here called Koelerioid Clades A and B. Phylogenetic relationships among koelerioid grasses are investigated using plastid DNA sequences of rpl32‐trnL, rps16‐trnK, rps16 intron, and ITS regions, focusing on Trisetum, Acrospelion, and some annual species (Rostraria p.p. and Trisetaria p.p.) closely related to Trisetum flavescens in Koelerioid Clade A. Phylogenetic analyses of several selected data sets performed for 80 taxa and using maximum likelihood and Bayesian methods, revealed mostly congruent topologies in the nuclear and plastid trees, but also reticulation affecting several lineages. Trisetum is restricted to one species, T. flavescens, which is a sister to the clade formed by Trisetum gracile and Trisetaria aurea. The latter two species are classified here in the genus Graciliotrisetum gen. nov. The sister clade includes three species of Rostraria and Trisetaria lapalmae, all of which are classified here in a resurrected genus, Aegialina, which includes four species. Acrospelion is enlarged to include 13 species after the addition of other species formerly classified in Trisetum sect. Trisetum and T. sect. Acrospelion. We also transfer Trisetum ambiguum, Trisetum longiglume, and Koeleria mendocinensis to Graphephorum; and Helictotrichon delavayi to Tzveleviochloa, expanding these genera to eight and six species, respectively. We evaluate cases of reticulate evolution between Koelerioid Clades A and B and within Koelerioid Clade A, which probably gave rise to Graphephorum, Rostraria cristata, and Rostraria obtusiflora. Finally, we comment on polyploidy and biogeographic patterns in koelerioid grasses. We propose the following 26 new combinations: Acrospelion alpestre, Acrospelion altaicum, Acrospelion argenteum, Acrospelion bertolonii, Acrospelion buschianum, Acrospelion buschianum subsp. transcaucasicum, Acrospelion fuscum, Acrospelion laconicum, Acrospelion macrotrichum, Acrospelion rigidum, Acrospelion rigidum subsp. teberdense, Acrospelion tamonanteae, Acrospelion velutinum, Aegialina lapalmae, Aegialina pubescens, Aegialina pumila, Aegialina pumila subsp. fuscescens, Aegialina salzmannii, Aegialina salzmannii subsp. cossoniana, Graciliotrisetum aureum, Graciliotrisetum gracile, Graphephorum ambiguum, Graphephorum longiglume, Graphephorum mendocinense, Graphephorum orthochaetum, and Tzveleviochloa delavayi. Lectotypes are designated for the names Aegialitis tenuis, Aira melicoides, Avena aspera var. parviflora, Avena delavayi, Koeleria grisebachii var. mendocinensis, Koeleria pubescens subsp. cossoniana, Koeleria pumila, Koeleria salzmannii, Phalaris pubescens, Trisetum aureum, Trisetum cernuum, Trisetum fuscescens, Trisetum longiglume, and Trisetum wolfii; and we designate one neotype for Alopecurus litoreus.
Phylogenetic analysis of Koeleriinae clades A and B yielded mostly congruent topologies in nuclear and plastid trees. However, several lineages resolved in strikingly incongruent positions in trees from different data sets, suggesting a reticulate origin for these taxa. Twenty-six new combinations and 14 lectotypes and one neotype were designated.