Jingjing Zhang, Yisi Hu, Lin Yang, Zhiwei Zhang, Shichao Wei, Wen Yu, Hao Luo, Fuwen Wei, Wenliang Zhou
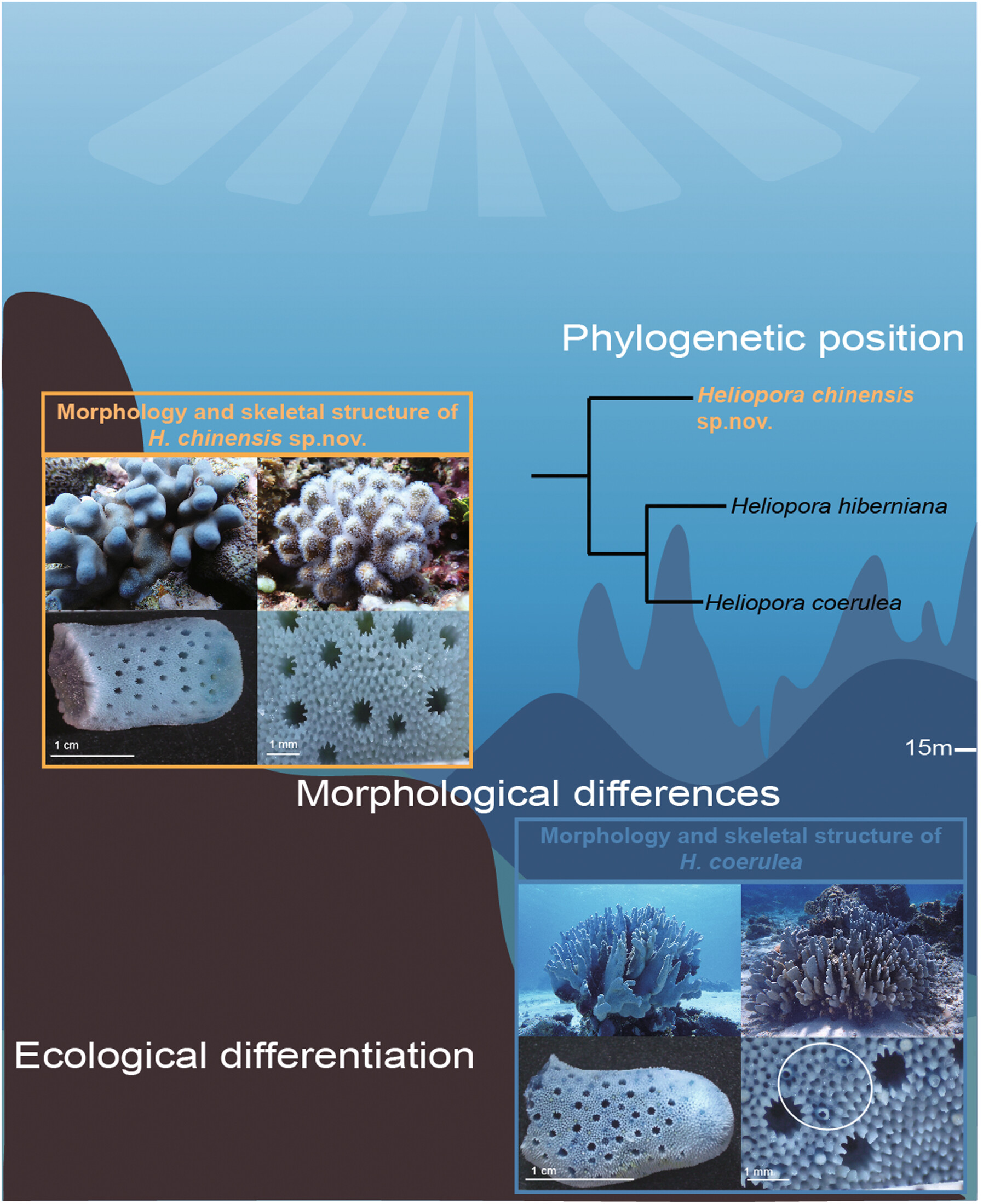
Heliopora (subclass Octocorallia, order Scleralcyonacea, family Helioporidae), commonly known as the “blue coral,” represents the only reef-building lineage within Octocorallia. The genus shows diverse growth forms of branching, encrusting, and laminar types, which leads to ambiguities in traditional morphology-based taxonomy. Here, we investigated the diversity of Heliopora species in the South China Sea (SCS) and their phylogenetic relationships across the Indo–Pacific using integrated morphological and phylogenomic approaches. Whole-genome resequencing of 52 colonies from the SCS islands, combined with published data from 244 samples worldwide, revealed three distinct clades: H. coerulea, H. hiberniana, and a previously undescribed lineage. Morphological analyses characterized the new lineage with a blue skeleton, a short columnar to encrusting growth form, large autopores with 12–15 pseudosepta, absence of worm tubes, and elaborated coenchymal echinulations. These features contrast with the long-branching to lobate H. coerulea and the white-skeletoned H. hiberniana. Based on its unique morphology and distinct phylogenetic position, we describe this lineage as a new species: Heliopora chinensis sp. nov. It is distributed mainly in the SCS islands, Taiwan of China, and the Ryukyu Islands. Meanwhile, global research and citizen science records suggest that H. hiberniana is restricted to the lower latitudes of Indo–Pacific Ocean, whereas H. coerulea occurs broadly across the Indo–Pacific. Our findings highlight the effectiveness of integrating phylogenomics and morphology to resolve coral systematics, uncover cryptic species diversity, and provide new insights into speciation, diversification, and conservation of corals, thus providing a critical taxonomic basis for informing future conservation strategies for coral reef ecosystems.
Integrating morphological, phylogenomic, and ecological evidence delimited species boundaries within the coral genus Heliopora. Whole-genome resequencing of 52 SCS colonies and 244 global genomes recovered three clades that show morphological, ecological, and genetic divergence, revealing cryptic diversity and strengthening the taxonomic basis for coral speciation and conservation strategies.