Francisco Fajardo‐Gutiérrez, Mariasole Calbi, Markus S. Dillenberger, Sebastian Tello, AlfredoFuentes, Nora H. Oleas, Ricardo A. Segovia, Christine E. Edwards, Yohan Pillon, James E.Richardson, Thomas Borsch
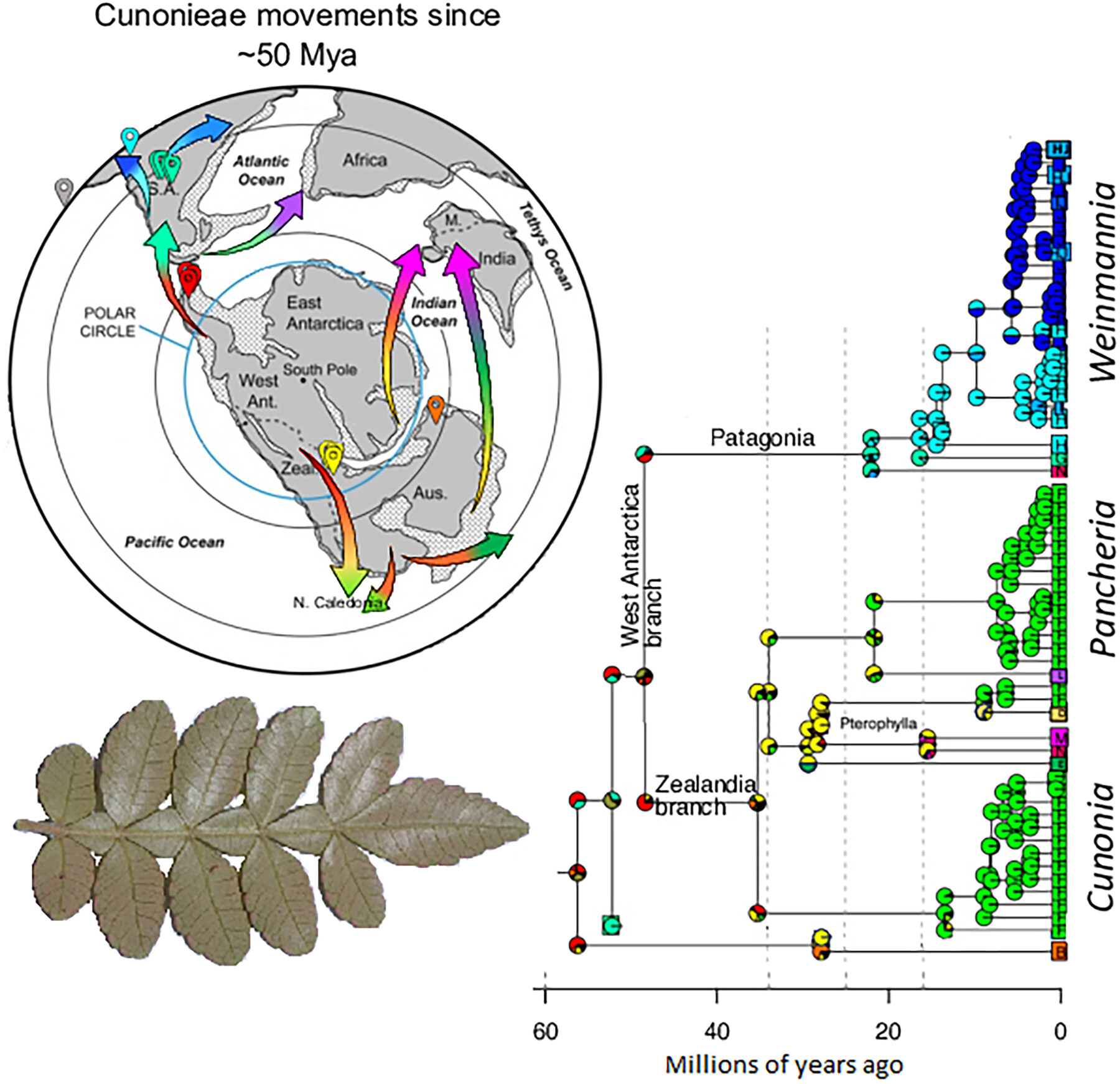
The tribe Cunonieae comprises five genera and 214 species of shrubs and trees currently distributed in the Southern Hemisphere and the tropics, exhibiting an amphi‐Pacific disjunct distribution shared with Araucariaceae, Myrtaceae, Nothofagaceae, Podocarpaceae, and Proteaceae, among others. To address the central question of how historical geological forces have shaped the distribution of plant diversity in the southern hemisphere, we aimed to provide evidence from the biogeographical history of Cunonieae. We generated themost densely sampled phylogenetic trees of Cunonieae available to date, with 121 samples and 81 species, basedon 404 new sequences of plastid and nuclear DNA regions with high hierarchical phylogenetic signal (matK, trnL‐F, rpl16, and internal transcribed spacer (ITS)). We included 184 samples of Rosids to estimate divergence times using fossil calibration points. For biogeographic inference, we employed a time‐stratified model including fossils as tips. Cunonia and Pterophylla were paraphyletic in the ITS tree, and Cunonia was paraphyletic in the plastid tree. Pancheria, Vesselowskya, and Weinmannia were monophyletic, the latter with conflicting nuclear and plastid phylogenies. The crown group Cunonieae was dated at ~56 Ma, and its ancestral areas were Antarctica and Patagonia. Antarctica acted as a bridge between Australia and South America before the consolidation of the Antarctic Ice Sheet and the extinction of the lineage in Antarctica from the Oligocene to the Miocene. Following that, Cunonieae spread to lower latitudes via Zealandia/Oceania and Patagonia/South America. Geological changes during the Pliocene facilitated a further burst in diversification along the Andes, in Madagascar, and in New Caledonia, where at least three colonization events occurred.
Fossil calibration reveals recent radiations and biogeographical history of the Cunonieae tribe. The most densely sampled phylogeny indicated Antarctica and Patagonia as areas of origin, confirming the reestablishment of Pterophylla and revealing the northward movement of Weinmannia into the tropical Andes. Conflicting plastid and nuclear phylogenies suggest past chloroplast capture.