Mario Rincón-Barrado, Sanna Olsson, Tamara Villaverde, Belén Moncalvillo, Lisa Pokorny, Alan Forrest, Ricarda Riina, and Isabel Sanmartín
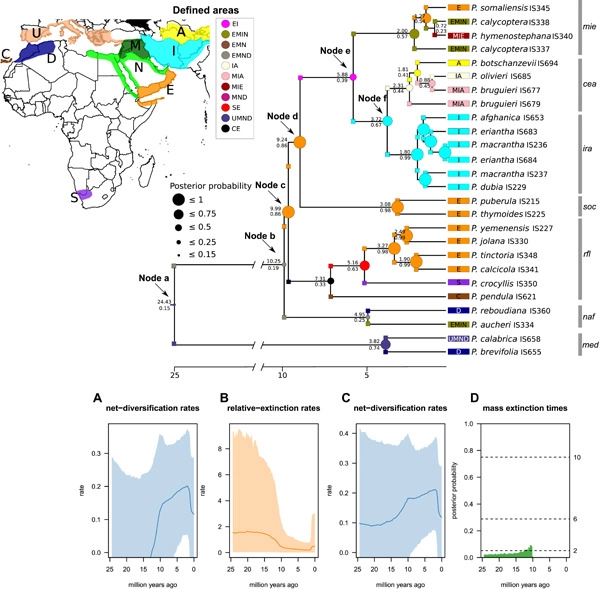
Wide-range geographically discontinuous distributions have long intrigued scientists. We explore the role of ecology, geology, and dispersal in the formation of these large-scale disjunctions, using the angiosperm tribe Putorieae (Rubiaceae) as a case study. From DNA sequences of nuclear ITS and six plastid markers, we inferred a phylogeny with 65% of all known Putorieae species. Divergence times, ancestral ranges, and diversification rate shifts were then estimated using Bayesian inference. We further explored species climatic tolerances and performed ancestral niche reconstruction to discriminate among alternative speciation modes, including geographical and ecological vicariance, and ecogeographical, ecological, and dispersal-mediated speciation. As a result, we identified seven major clades in Putorieae, some of which exhibit striking geographical disjunctions, matching the Rand Flora pattern, with sister species in the Canary Islands and eastern and southern Africa. Initial diversification within the tribe occurred in the early Miocene, coincident with a period of climate warming; however, most clades diverged within the last 10 Myr. Aridification and high extinction rates, coupled with ecological vicariance, explain the oldest disjunctions. Adaptation to new environmental conditions, after allopatry, is observed in several clades. Dispersal, either long-distance or via corridors made available by mountain uplift, is behind the most recent disjunctions. Some of these events were followed by ecological speciation and rapid diversification, with species becoming adapted to xeric or increasingly colder continental climates. We show that an integrative approach may help discriminate among speciation modes invoked to explain disjunctions at macroevolutionary time scales, even when extinction has erased the signature of past events.
Wide-range geographically discontinuous distributions have long intrigued scientists. We explore the role of ecology, geology, and dispersal in the formation of these large-scale disjunctions, using the angiosperm tribe Putorieae (Rubiaceae) as a case study. From DNA sequences of nuclear ITS and six plastid markers, we inferred a phylogeny, obtained an ancestral niche reconstruction, and estimated divergence times, ancestral ranges, and diversification rate shifts using Bayesian inference. As a result, we identified seven major clades in Putorieae, some of which exhibit striking geographical disjunctions and a probable reason for their distribution, including ecological and geographical vicariance, ecological and ecogeographical speciation, and long-distance dispersal.