


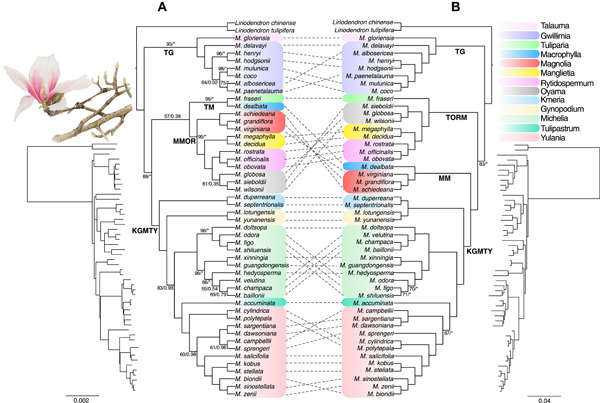
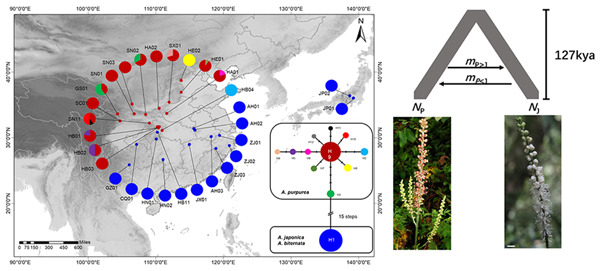
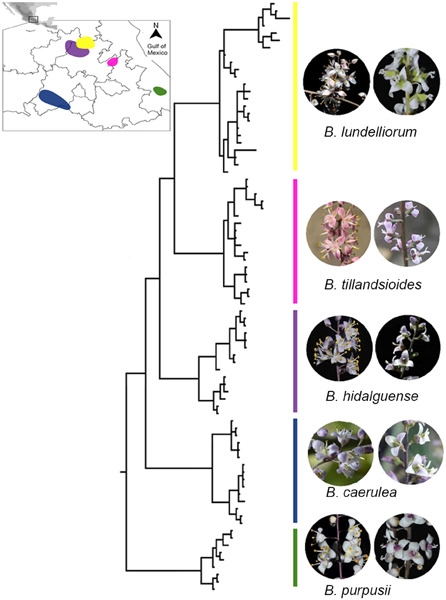
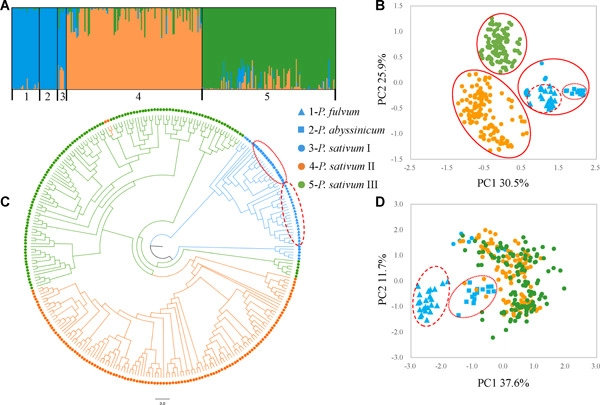
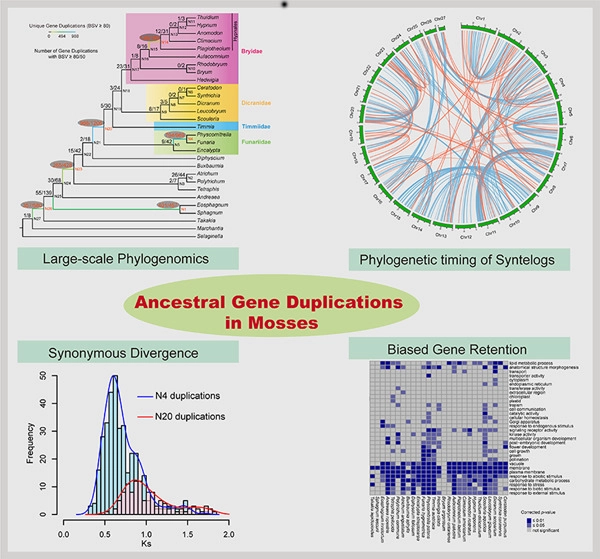
Mosses (Bryophyta) are a key group occupying an important phylogenetic position in land plant (embryophyte) evolution. The class Bryopsida represents the most diversified lineage, containing more than 95% of modern mosses, whereas other classes are species‐poor. Two branches with large numbers of gene duplications were elucidated by phylogenomic analyses, one in the ancestry of all mosses and another before the separation of the Bryopsida, Polytrichopsida, and Tetraphidopsida. The analysis of the phylogenetic progression of duplicated paralogs retained on genomic syntenic regions in the Physcomitrella patens genome confirmed that the whole‐genome duplication events WGD1 and WGD2 were re‐recognized as the ψ event and the Funarioideae duplication event, respectively. The ψ polyploidy event was tightly associated with the early diversification of Bryopsida, in the ancestor of Bryidae, Dicranidae, Timmiidae, and Funariidae. Together, four branches with large numbers of gene duplications were unveiled in the evolutionary past of P. patens. Gene retention patterns following the four large‐scale duplications in different moss lineages were analyzed and discussed. Recurrent significant retention of stress‐related genes may have contributed to their adaption to distinct ecological environments and the evolutionary success of this early‐diverging land plant lineage.

The Journal of Systematics and Evolution would like to acknowledge and thank the following reviewers for their contributions in the period January 1–December 31 in 2021:
Allen, Geraldine
Appelhans, Marc
Areces-Berazain, Fabiola
Averianov, Alexander O.
Bai, Wei-Ning
Bard, Nicholas
Bell, Charles
Besnard, Guillaume
Bisang, Irene
Bitencourt, Cássia
Bouckaert, Remco
Brandrud, Marie Kristine
Buellesbach, Jan
Câmara, Paulo
Cameron, Kenneth
Cannon, Chuck
Carta, Angelino
Chen, Jun
Chen, Xiao-Yong
Chen, Zhi-Duan
Christie, Kyle
Corlett, Richard
Dering, Monika
Ding, Wen-Na
Durka, Walter
Ebersbach, Jana
Edwards, Christine
Engle-Wrye, Nick
Eriksson, Torsten
Escudero, Marcial
Fady, Bruno
Favre, Adrien
Fazekas, Aron
Ford, Kerry
Franco, Fernando
Gao, Jiang-Yun
Gao, Lian-Ming
Gauquelin, Thierry
Ge, De-Yan
Ge, Xue-Jun
Gerrath, Jean
Goertzen, Leslie
Gong, Yan-Bing
Gradstein, Robbert
Harris, AJ
Hoffmann, Matthias H.
Howard, Cody
Huang, Ji-Hong
Huang, Jia-Xing
Huang, Shuang-Quan
Huarte, Roberto
Hultine, Kevin R.
Ickert-Bond, Stefanie
Ignatov, Michael
Ikeda, Hajime
Jiang, Hong-En
Jiménez-Mejías, Pedro
Jin, Xiao-Hua
Johnson, Steven D.
Kainulainen, Kent
Kandasamy, Dineshkumar
Kang, Long-Li
Kang, Ming
Klahs, Phillip
Klopper, Ronell
Kooyman, Robert
Ksepka, Daniel
Kutanan, Wibhu
Landrein, Sven
Lepais, Olivier
Li, Bo
Li, Jianhua
Li, Lin-Feng
Li, Xin-Xin
Li, Zhong-Hu
Liang, Er-Yuan
Liao, Wan-Jin
Liu, Bo-Ling
Liu, Xiao-Yan
Liu, Zhi-Jin
Liu, Zhi-Yong
Lou, Hai-Yi
Luo, A-Rong
Luo, Yi-Bo
Ma, Liang
Ma, Yong-Peng
Mao, Jian-Feng
Mao, Kang-Shan
Márquez-Corro, José Ignacio
Martín-Bravo, Santiago
McFarlane, Terry
McNeal, Joel
Michelangeli, Fabian A.
Miller, Joseph
Mondal, Mayukh
Morgan, John
Musser, Grace
Nawal, Shrestha
Oh, Sang-Hun
Olofsson, Jill
Ortiz, Edgardo
Passalacqua, Nicodemo
Pennell, Matthew
Phillips, Matthew
Pipes, Leonore
Pócs, Tamás
Qian, Hong
Qiu, Ying-Xiong
Ranker, Tom
Razafimandimbison, Sylvain G.
Rebollo, Roberto
Ree, Richard
Ren, Guang-Peng
Ren, Zong-Xin
Roalson, Eric
Roeser, Martin
Ruiz-Sanchez, Eduardo
Saarela, Jeffery
Salazar-Mendias, Carlos
Sánchez-Vilas, Julia
Sanmartin, Isabel
Santos-Gally, Rocio
Schlueter, Philipp
Schneider, Harald
Schwery, Orlando
Shrestha, Nawal
Shi, Tao
Šmarda, Petr
Sniderman, Kale
Snow, Neil
Song, Gang
Song, Yi-Gang
Soreng, Robert
Sosa, Pedro A.
Stark, Lloyd
Stedje, Brita
Su, Tao
Su, Xu
Sun, Hang
Sun, Kun
Sun, Miao
Sun, Yan-Xia
Sun, Yong-Shuai
Sundue, Michael
Thiv, Mike
Thomas, Daniel
Tian, Bao-Liang
Tribble, Carrie
Triplett, Jimmy
Vaillancourt, Rene
Vallès, Joan
van der Merwe, Marlien
Vanderpoorten, Alain
Villar, Jose
von Balthazar, Maria
Vorontsova, Maria S.
Walker, Joseph F.
Wang, Bao-Sheng
Wang, Heng-Chang
Wang, Hongru
Wang, Jing
Wilson, Paul
Wu, Yong-Jie
Wu, Zhi-Qiang
Xiang, Chun-Lei
Xiang, Qiao-Ping
Xie, Shu-Lian
Xing, Yao-Wu
Xu, Qing
Xu, Xiao-Ting
Yan, Yujing
Yang, Shi-Xiong
Yang, Ying-Bo
Yu, Yan
Zander, Richard
Zhang, De-Zhi
Zhang, Xiao-Ming
Zhang, Zhi-Yong
Zhao, Shi-Lei
Zhou, Ren-Chao
Zu, Pengjuan












