


The JSE established Awards of JSE Outstanding Papers in 2014 and has awarded two Outstanding Papers and two Outstanding Papers by Young Investigators each year since 2008 (Ge & Wen, 2015). The selection of the awards was based on votes and assessments from all 17 editors of the journal with the consideration of citation data from Web of Science and the impact on systematics and evolution. The winners of the Awards receive a certificate and a prize of $800 (JSE Outstanding Papers) or $500 (JSE Outstanding Papers by Young Investigators). Here, we are delighted to announce the four winners of the awards selected from JSE publications in 2020 and highlight the significance of these papers.
As an early diverged lineage within the Magnoliids, the family Magnoliaceae is critical in systematic position on the Tree of Life and consists of many species with important values in timber production and traditional medicines, in addition to being ornamental plants. Nevertheless, the taxonomy and particularly the delimitations of genera and/or sections in this family have been highly controversial among taxonomists, with as few as two and as many as 17 genera. Wang et al. (2020) reconstructed the phylogeny of Magnoliaceae using sequences of the complete chloroplast genomes with a broad taxon sampling of 86 species. In conjunction with morphological and geographic evidence, they recognized two subfamilies Liriodendroideae and Magnolioideae, each with one genus (Liriodendron and Magnolia, respectively). Specifically, their results strongly supported 15 major clades within Magnolia s. l., inconsistent with the previous subgeneric treatment that recognized three subgenera. The authors further detected the data incongruence among several major clades and discussed the generic delimitation and phylogenetic relationships within the family. Overall, this work helps establish a better classification of Magnoliaceae and provides new insights into the global biogeographic diversification of this family with both temporal and tropical elements.
How plants adapt to globe climate change matters both for survival and extinction of plants and for agricultural and environmental sustainability and food security. In this review, Anderson & Song (2020) addressed (i) whether climate change exerted novel selection and disrupted local adaptation, (ii) how gene flow facilitated adaptive responses to climate change, and (iii) whether adaptive phenotypic plasticity could sustain populations in the short term. They also reviewed studies that tested the influence of climate change on species interactions, predicted the adaptive potential of plants under climate change, and dissected the genetic basis of plant adaptation to climate change. By highlighting several key research gaps, the authors encouraged additional applications of emerging genomic tools, along with interdisciplinary investigations, to enhance our ability to predict the adaptive potential of plants under climate change and to elucidate the genetic basis of complex trait variation.
The family Lauraceae is a major component of tropical and subtropical forests worldwide, and includes many species that provide important economic products, including timber, perfume, spices, herbal medicines, and fruit crops. However, phylogenetic relationships within Lauraceae remain unsolved because of various reasons. Song et al. (2020) compiled a large data set that includes 43 newly sequenced and 77 downloaded plastomes representing 42 genera of Lauraceae and 17 related families of angiosperms. On this basis, they reconstructed the phylogenetic relationships within the Lauraceae and among seven of the nine families of the Laurales. In combination with the morphological evidence, the authors confirmed the monophyly of Lauraceae and identified nine monotypic clades that offered insights to improve the tribal classification of Lauraceae. They also described two new tribes (Caryodaphnopsideae and Neocinnamomeae) and updated the compositions of four other tribes. This study provides a robust phylogenetic framework through which to address the evolutionary history of the Magnoliids, the third-largest group of Mesangiospermae.
The uneven distribution of taxonomic diversity among the branches of the Tree of Life and geographic regions is one of the most intriguing puzzles in biology and has been hypothesized to arise from significant differences in the parameters governing rates of speciation and extinction. To address this issue, Echeverría-Londoño et al. (2020) assembled a set of time-calibrated and species-level phylogenies of extant Solanum species, including 1169 of its 1234 species, to reconstruct diversification rates across lineages and analyze them in a geographical context. They (i) explored the origins of the high heterogeneity of species richness among subclades of this genus, (ii) investigated the relative importance of clade-specific, tree-wide, and geographic variation in evolutionary rates, and (iii) analyzed how these patterns were associated with historical biogeographic movements of lineages and/or environmental changes. The results showed that the lineages in the Old World were diversifying more rapidly, which coincided with a long-distance dispersal event from the Neotropics to regions where major climatic changes were taking place. In addition, two separate groups of Solanum have migrated and established in Australia, with only the arid adapted lineages being significantly increased in diversification rate. Together, these findings provide a clear example of how successful colonization of new areas and niches could drive explosive diversifications.
We want to congratulate the winners of the JSE Awards for their important contributions to systematics and evolution! We cordially invite the many colleagues in systematics and evolution to submit their first-rate research to JSE in the coming years.
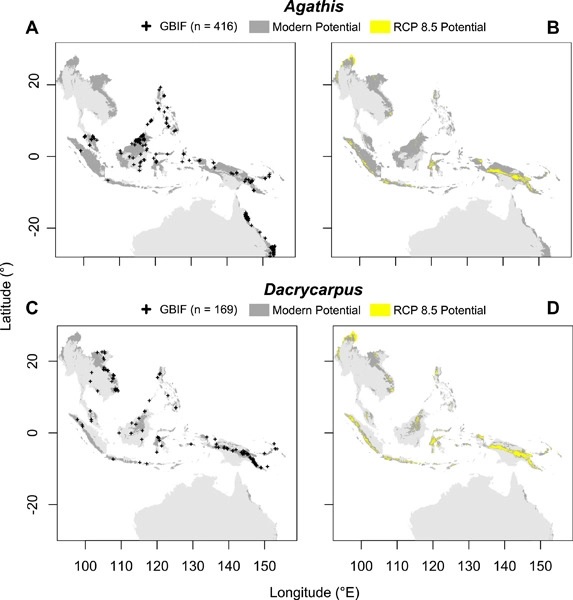
The Southeast Asian rainforest region is extremely complex and biodiverse. Fossils have shown that paleo-Antarctic rainforest lineages (PARLs) now extant in Asia tracked the ever-wet conditions needed to survive and diversify through deep time. However, the threat of future climate change to the remaining rainforest and PARLs in Southeast Asia has yet to be evaluated to set conservation priorities. We first quantified the woody-genus floristic relationships of Southeast Asian Island Groups by vetting and analyzing recent compilations of bioregional species data. We then evaluated the contributions to community assembly of woody fossil lineages and Island Group relationships to environmental gradients. To better understand climatic constraints of fossil lineage distributions and forecast distributions under projected future climate, we used exemplar living woody PARLs, including two angiosperms and two gymnosperms. Generalized linear models were used to project potential distributions under future climate pathways that assume no reduction in carbon dioxide emissions. The floristic analyses highlighted strong similarities among Island Groups in the ever-wet forest areas of Malesia, where PARLs are often concentrated. Ordination outliers represented more seasonal locations. Species distribution models showed that potential future distributions of ancient lineages are constrained by increasing rainfall seasonality and higher seasonal temperatures, with significant differences among exemplar genera. Notably, potential distributions often mapped onto de facto inaccessible areas, where forest clearing and the ubiquitous marine dispersal barriers that characterize the region will drastically inhibit potential relocation. These realities gravely threaten paleo-conservation values and contemporary rainforest community assembly processes in Southeast Asia.
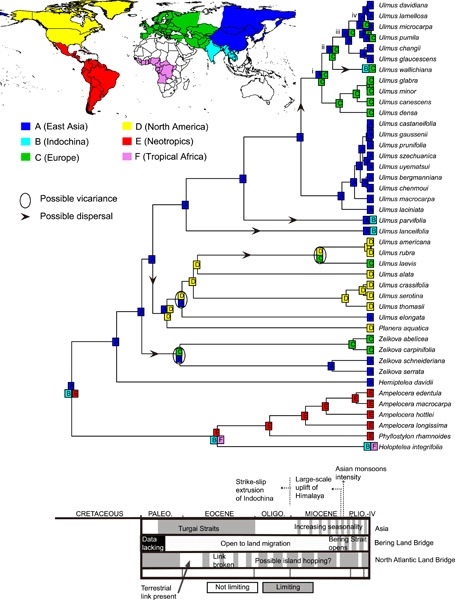
Ulmaceae is a woody family widespread in northern temperate forests. Despite the ecological importance of this family, its phylogeny and biogeographic history are poorly understood. In this study, we reconstruct phylogenetic relationships within the family and infer spatio-temporal diversification patterns based on chloroplast genome (complete cpDNA) and nuclear ribosomal DNA sequences (nrDNA). The seven Ulmaceae genera are resolved in two main clades (temperate vs. tropical) by both cpDNA and nrDNA sequences. The temperate clade includes four genera, Hemiptelea, Zelkova, Planera, and Ulmus. The relationships among Planera and other genera are controversial because of inconsistent topologies between plastid and nuclear data. The tropical clade includes three genera ((Ampelocera, Phyllostylon), Holoptelea). Molecular dating and diversification analyses show that Ulmaceae originated in the Early Cretaceous (ca. 110–125 Ma) with the main lineages establishing from the Late Cretaceous to the early Eocene. The diversification rate slowed during the middle to the late Paleogene (ca. 23–45 Ma), followed by a rapid diversification of the East Asian temperate group in the Neogene, congruent with a global cooling event. The ancestral state optimization analysis suggests an East Asian origin of the temperate Ulmaceae clade during the Paleocene, which is consistent with the fossil record. Both phylogenomic and fossil evidence support East Asia as a center of origin and diversification for the temperate woody lineages.
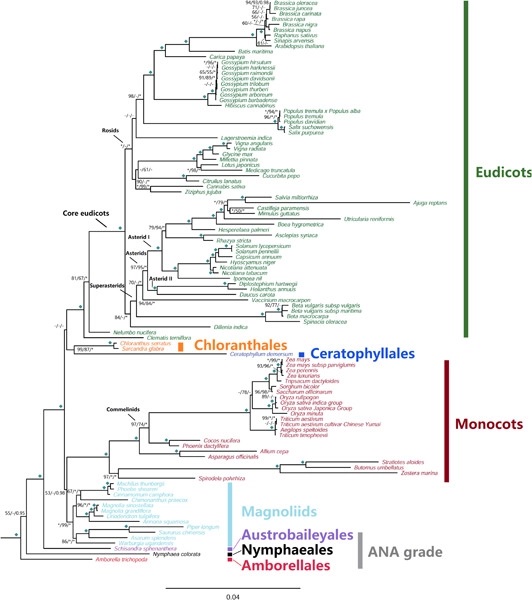
The early diversification of angiosperms is thought to have been a rapid process, which may complicate phylogenetic analyses of early angiosperm relationships. Plastid and nuclear phylogenomic studies have raised several conflicting hypotheses regarding overall angiosperm phylogeny, but mitochondrial genomes have been largely ignored as a relevant source of information. Here we sequenced mitochondrial genomes from 18 angiosperms to fill taxon-sampling gaps in Austrobaileyales, magnoliids, Chloranthales, Ceratophyllales, and major lineages of eudicots and monocots. We assembled a data matrix of 38 mitochondrial genes from 107 taxa to assess how well mitochondrial genomic data address current uncertainties in angiosperm relationships. Although we recovered conflicting phylogenies based on different data sets and analytical methods, we also observed congruence regarding deep relationships of several major angiosperm lineages: Chloranthales were always inferred to be the sister group of Ceratophyllales, Austrobaileyales to mesangiosperms, and the unplaced Dilleniales was consistently resolved as the sister to superasterids. Substitutional saturation, GC compositional heterogeneity, and codon-usage bias are possible reasons for the noise/conflict that may impact phylogenetic reconstruction; and angiosperm mitochondrial genes may not be substantially affected by these factors. The third codon positions of the mitochondrial genes appear to contain more parsimony-informative sites than the first and second codon positions, and therefore produced better resolved phylogenetic relationships with generally strong support. The relationships among these major lineages remain incompletely resolved, perhaps as a result of the rapidity of early radiations. Nevertheless, data from mitochondrial genomes provide additional evidence and alternative hypotheses for exploring the early evolution and diversification of the angiosperms.

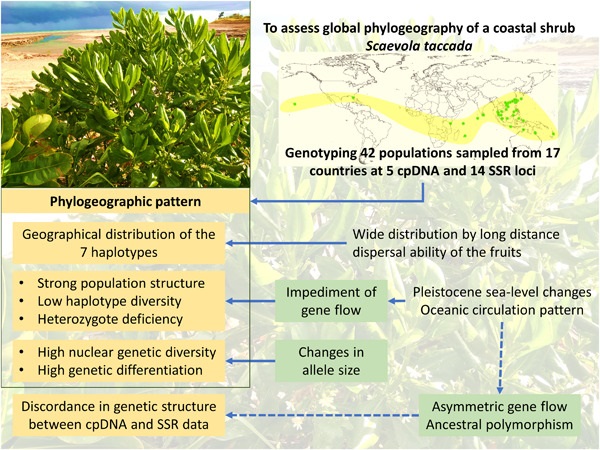
The phylogeography of coastal plant species is heavily influenced by past sealevel fluctuations, dispersal barriers, and life-history traits, such as long-distance dispersal ability of the propagules. Unlike the widely studied mangroves, phylogeographic patterns have remained mostly obscure for other coastal plant species. In this study, we sampled 42 populations of Scaevola taccada (Gaertn.) Roxb., a coastal shrub of the family Goodeniaceae, from 17 countries across its distribution range. We used five chloroplast DNA (cpDNA) and 14 nuclear microsatellite (simple sequence repeat [SSR]) markers to assess the influence of abiotic factors and population genetic processes on the phylogeographic pattern of the species. Geographical distribution of cpDNA haplotypes suggests that the species originated in Australia, followed by historical dispersal and expansion of its geographic range. Multiple abiotic factors, including the sealevel changes during the Pleistocene, the presence of landmasses like the Malay Peninsula, and contemporary oceanic circulation patterns, restricted gene flow between geographically distinct populations, thereby creating low haplotype diversity and a strong population structure. Population genetic processes acted on these isolated populations, leading to high nuclear genetic diversity and population differentiation, as revealed from analyzing the polymorphic SSR loci. Although genetic divergence was mostly concordant between cpDNA and SSR data, asymmetrical gene flow and ancestral polymorphism could explain the discordance in the detailed genetic structure. Overall, our findings indicate that abiotic factors and population genetic processes interactively influenced the evolutionary history and current phylogeographic pattern of S. taccada across its distribution range.
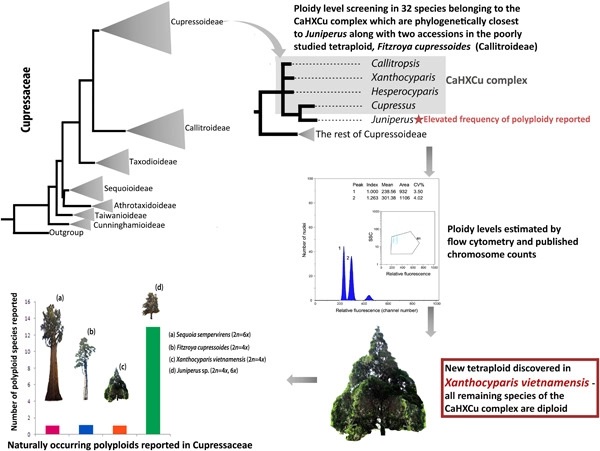
While polyploidy (whole-genome multiplication) is generally considered rare in extant gymnosperms (with the exception of Ephedra, Ephedraceae), the occurrence of sporadic polyploid individuals belonging to various genera in the conifer family Cupressaceae has been reported in the literature. In addition, recent studies have revealed that polyploidy is not uncommon in the genus Juniperus (Cupressaceae), with tetraploid and hexaploid individuals reported in individuals collected from wild populations. Given these findings, we undertook a comprehensive screening of ploidy levels in 32 species belonging to the four genera that are phylogenetically closest to Juniperus (i.e., Callitropsis, Hesperocyparis, Xanthocyparis, and Cupressus), referred to as the CaHXCu complex. In addition, we also determined the ploidy level of two accessions in the poorly studied tetraploid, Fitzroya cupressoides. Using flow cytometry together with published chromosome counts to assign ploidy levels, we show that all species of the CaHXCu complex are diploid except Xanthocyparis vietnamensis, which is tetraploid, with a genome size of 44.60 pg/2 C. This study opens up new opportunities for studying the impact and consequences of polyploidy on the evolution and adaptation of species in Cupressaceae.
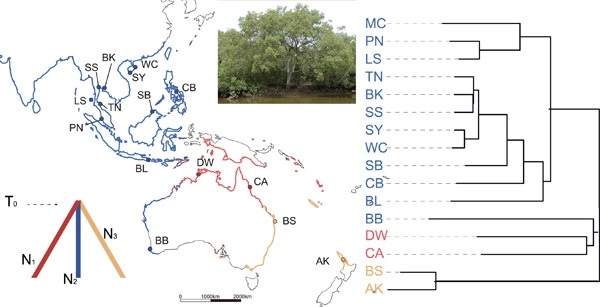
Subspecies is used to designate taxa below species but above geographical populations. What patterns of genomic variation are expected if taxa are designated as subspecies? In this study, we carry out such a survey on the mangrove tree Avicennia marina (Forssk.) Vierh. of the Indo-West Pacific coasts. This species has three subspecies, distinguished by morphological traits and geographical distribution. We collected samples from 16 populations (577 individuals) covering all three subspecies and sequenced 94 nuclear genes. We reveal comprehensive genetic divergence among subspecies, generally higher than among geographical populations within subspecies. The level of genetic diversity differs among the three subspecies, possibly hinting at a degree of separation among their gene pools. We observed that divergence varies from locus to locus across the genome. A small portion of the genome is most informative about subspecies delineation, whereas the rest is undifferentiated or slightly differentiated, hinting at uneven gene flow and incomplete isolation. The three subspecies likely split simultaneously with gene flow among lineages. This reticulate evolution results in some discordance between morphology and genetics in areas of population contact. In short, A. marina subspecies show species-like patterns in some respects and population-like patterns in others. We propose that the subspecies designated in A. marina are informative in predicting genetic divergences and useful in making conservation decisions.
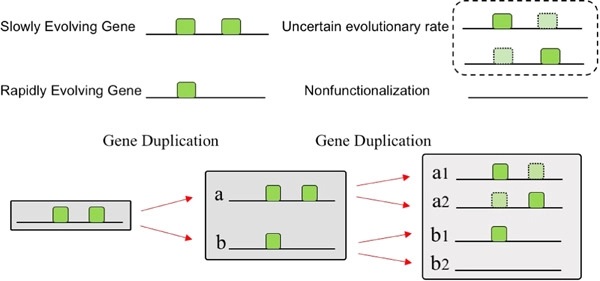
Gene duplication provides raw material for functional innovation, but gene duplicability varies considerably. Previous studies have found widespread asymmetrical sequence evolution between paralogs. However, it remains unknown whether the rate of evolution among paralogs affects their propensity of being retained after another round of whole-genome duplication (WGD). In this study, we investigated gene groups that have experienced two successive WGDs to determine which of two older duplicates with different evolutionary rates was more likely to retain both younger duplicates. To uncouple the measurement of evolutionary rates from any assignment of duplicate or singleton status, we measured the evolutionary rates of singleton genes in out-lineages but classified these singleton genes according to whether they are retained or not in a crown group of species. We found that genes that retained younger duplicates in the crown group of genomes were more constrained prior to the younger duplication event than those that failed to leave duplicates. In addition, we also found that the retained clades have more genes in out-lineages. Subsequent analyses showed that genes in the retained clades were expressed more broadly and highly than genes in the singleton clades. We concluded that the set of repeatedly retained genes after two WGDs is biased toward slowly evolving genes in angiosperms, suggesting that the potential of genes for both functional conservation and divergence likely affects their propensity of being retained after WGD in angiosperms.

An accurate understanding of species diversity is essential to studies across a wide range of biological subdisciplines. However, species delimitation remains challenging in evolutionary radiations, particularly in those herbaceous plants associated with microendemic, naturally fragmented distribution systems, where genotypic and phenotypic traits likely evolved discordantly. The Primula merrilliana complex, which is endemic to eastern China and has high horticultural value, used to be treated as one species but several clues suggested it might be composed of multiple species. Here we used multiple lines of evidence, including molecular, morphological, reproductive isolation, and geographic data, to assess independently evolving lineages within this complex. Our results indicated that the species diversity in the complex was underestimated previously, and four species (independently evolving lineages) can be recognized, including two new species described here. The extensive variation of the breeding system, especially the floral morph transition from distyled (outcrossing) to homostyled (selfing) multiple times, possibly promoted the rapid speciation within such a small geographic scale. This study case indicated that the phenomenon of genetically highly divergent but morphologically indistinguishable is perhaps shown in herbs with fragmented distributions; the alternative extreme evolutionary phenomenon, in which complete reproductive barriers have been accumulated but with little genetic differentiation, also exists. Thus we highlight the importance of incorporating other characters, such as postzygotic reproductive isolation and geographic data, with commonly used molecular and morphological traits to infer species boundaries through an integrative taxonomic approach in such systems.
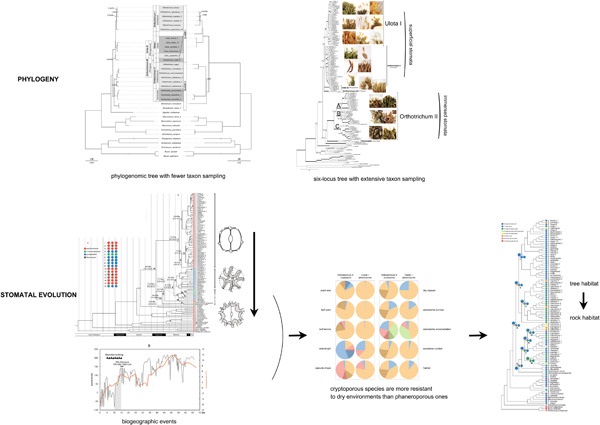
Orthotrichum Hedw. s.l. and Ulota Mohr s.l. are two of the most speciose genera of the xerophytic moss family Orthotrichaceae. We reconstructed the phylogeny of these two genera using three data matrices: (i) organellar genomes and 33 taxa; (ii) six loci from three genomes and 144 taxa; and (iii) two plastid loci and 163 taxa. The present phylogeny, based on the maximum sampling of genes or taxa to date, generally confirms the new classification of Orthotrichum and Ulota, and indicated that all Ulota species, except Ulota phyllantha Brid., form a clade and three lineages comprise the cryptoporous Orthotrichum clade. We provided new morphological characters that support the present division of the two genera. Ancestral state reconstruction of stoma indicates that superficial stomata in Orthotrichum represent a plesiomorphic character and semi-immersed stomata were derived from immersed stomata. The results also suggest that immersed stomata independently arose once in Orthotrichum, whereas semi-immersed stomata probably arose more than once. Molecular dating analysis reveals that the occurrence of immersed stomata is probably related to arid environments during the early Oligocene to late Miocene, whereas the appearance of semi-immersed stomata might be associated with the mesic–xeric or semiarid environments during the middle Miocene to Pliocene. Ancestral state reconstruction of habitat indicates that the saxicolous habitat is apomorphic and independently evolved multiple times in Orthotrichum and Ulota, which supports the former hypothesis. Considering morphological statistics, the development of the cryptopore in Orthotrichum could provide increased resilience to dry habitats, and might promote their habitat shift during evolution.

Stipa shanxiensis, a cryptic species within Stipa grandis that originated from central and western China, is described based on morphological, genomic, and ecological data from field and common garden experiments. Stipa shanxiensis morphologically resembles S. grandis, although phylogenetically it is closely related to the less morphologically similar Stipa baicalensis and Stipa krylovii. Of the eight significant morphological differences between S. shanxiensis and S. grandis, the two, cauline ligules longer than 2 cm with a filiform apex, and hairs shorter than 0.2 mm on the adaxial surface of the cauline uppermost leaves can be used to distinguish the species. Results from a common garden experiment verified that the two diagnostic characteristics were relatively stable and less morphologically plastic in response to environmental variation. Furthermore, a significant ecological divergence was found between S. shanxiensis and S. grandis, such that the former preferred warmer and more humid climates, and their predicted distribution was generally separated. Taken together, our results highlight that the integrative taxonomic approach was valuable for recognizing a new cryptic species in Stipa. In particular, we find that common garden experiments involving the effects of growth stage and characteristic position helped to morphologically diagnose cryptic species. These findings may also facilitate our understandings of ecological adaption and phenotypic plasticity in response to environmental change.
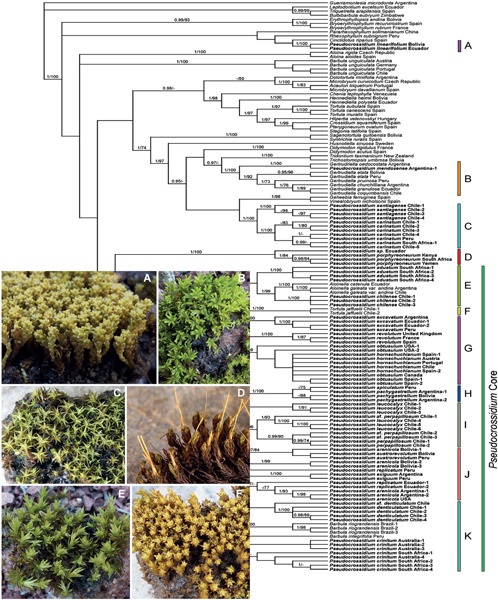
Pseudocrossidium is a genus of 21 species belonging to the Pottiaceae with the highest concentration of taxa and morphological variation found in South America. To investigate the evolutionary relationships among the species of Pseudocrossidium and other members of the Pottioideae, molecular phylogenetic reconstructions, using the nuclear ITS1-5.8S-ITS2, and the plastid atpB-rbcL, trnG, and trnL-F, has been performed because this genus has only been partially tested using molecular markers. Bayesian and maximum likelihood topologies show that the genus, as presently circumscribed, is polyphyletic. Consequently, the circumscription of Pseudocrossidium is amended and numerous taxonomic changes resulting from the molecular, morphological, and nomenclatural studies are proposed. The phylogenetic and morphologically divergent Pseudocrossidium mendozense is renamed as Gertrudiella mendozensis. Pseudocrossidium linearifolium and P. porphyreoneurum are representatives of the new genera Barbulastrum and Helicobarbula, respectively. Pseudocrossidium carinatum and P. santiagense are accommodated in a new genus Austrobarbula. Aloinella, nested in a paraphyletic Pseudocrossidium, is maintained at generic rank, apparently derived from Pseudocrossidium. Barbula integrifolia, B. riograndensis, and Tortula jaffuelii are transferred to Pseudocrossidium. The remaining species of Pseudocrossidium are maintained in this genus, pending further studies. Conflicts of the trees observed could be evidence of interspecific or intergeneric gene flow in various lineages in the Pottioideae.

Amber is renowned for the exceptional preservation state of its inclusions, allowing detailed morphological analysis and providing relevant environmental, palaeoecological, geographical, and geological information. Amber deposits are predominantly known from North America, Europe, and Asia, and are considered to be rare on the continents that formed Gondwana. The recent discovery of fossiliferous amber deposits in Ethiopia, therefore, provides an inimitable opportunity to close gaps in the fossil record of African terrestrial biota and to study organisms which are otherwise rare in the fossil record. Here we show that diverse cryptogams are preserved in highest fidelity in Miocene Ethiopian amber. We describe gametophyte fragments of four liverworts: Thysananthus aethiopicus sp. nov. (Porellales, Lejeuneaceae), Lejeunea abyssinicoides sp. nov. (Porellales, Lejeuneaceae), Frullania shewanensis sp. nov. (Porellales, Frullaniaceae), and Frullania palaeoafricana sp. nov. (Porellales, Frullaniaceae). Furthermore, we describe a pleurocarpous moss of the extant genus Isopterygium (Hypnales, Pylaisiadelphaceae) and a lichen representing the order Lecanorales. These new specimens represent the first amber fossils of liverworts, mosses, and lichens from the African continent and render Ethiopian amber as one of the few worldwide amber deposits preserving bryophytes (mosses and liverworts) or lichens. Fossil species of Thysananthus were recorded in Eocene Baltic and Oligocene Bitterfeld as well as Miocene Dominican and probably also Miocene Mexican ambers. Fossils that can unequivocally be assigned to Lejeunea have only been found in Dominican amber so far. Neotropical ambers contain only one taxon of Frullania to date, while the genus is most diverse in Baltic, Bitterfeld, and Rovno ambers, formed in temperate regions. The new fossils support a tropical to subtropical origin of Ethiopian amber. The new African liverwort fossils are included in an updated list of leafy liverworts described from worldwide Cenozoic ambers to date.
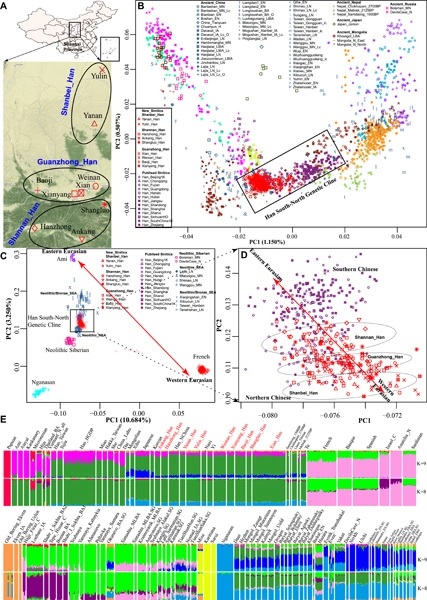
The Han Chinese are the world's largest ethnic group residing across China. Shaanxi province in northern China was a pastoral–agricultural interlacing region sensitive to climate change since Neolithic times, which makes it a vital place for studying population dynamics. However, genetic studies of Shaanxi Han are underrepresented due to the lack of high-density sampling and genome-wide data. Here, we genotyped 700 000 single nucleotide polymorphisms (SNPs) in 200 Han individuals from nine populations in Shaanxi and compared with available modern and ancient Eurasian individuals. We revealed a north–south genetic cline in Han Chinese with Shaanxi Han locating at the northern side of the cline. We detected the western Eurasian-related admixture in Shaanxi populations, especially in Guanzhong and Shanbei Han Chinese in proportions of 2%–4.6%. Shaanxi Han were suggested to derive a large part of ancestry (39%–69%) from a lineage that also contributed largely to ancient and present-day Tibetans (85%) as well as southern Han, supporting the common northern China origin of modern Sino-Tibetan-speaking populations and southwestward expansion of millet farmers from the middle-upper Yellow River Basin to the Tibetan Plateau and to southern China. The rest of the ancestry of Shaanxi Han was from a lineage closely related to ancient and present-day Austronesian and Tai-Kadai speaking populations in southern China and Southeast Asia. We also observed a genetic substructure in Shaanxi Han in terms of north–south-related ancestry corresponding well to the latitudes. Maternal mitochondrial DNA and paternal Y-chromosome lineages further demonstrated the aforementioned admixture pattern of Han Chinese in Shaanxi province.












