



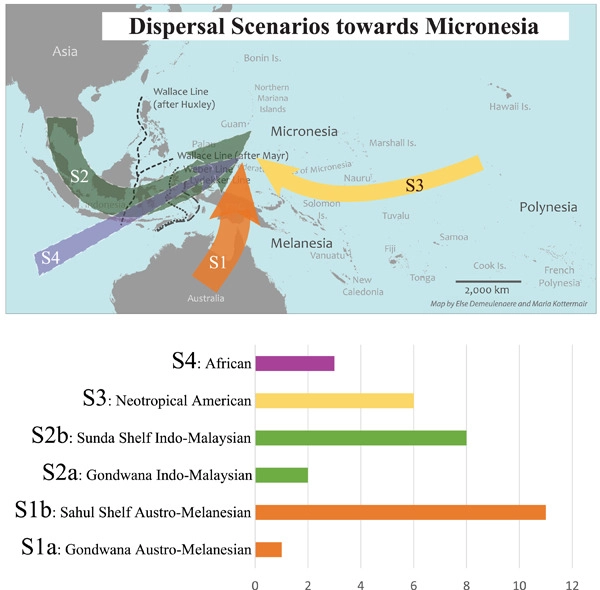
Micronesian islands taxa show high endemism rates, but very little is known about their biogeographical histories. The lack of systematic biogeography is mainly due to insufficient phylogenetic research in Micronesia. With the recent increase in published molecular biogeographic data, we were able to, for the first time, answer fundamental biogeography questions by reviewing and analyzing numerous geological, ecological, and evolutionary studies. This review, in addition to providing an overview of Micronesian geological history, confirmed the importance of long-distance dispersal mechanisms and founder-event speciation, and morphological and physiological adaptations of plant propagules to cross vast stretches of ocean by wind, ocean currents, bird, or bat dispersal. These adaptations to habitat and geological features, including reef types, determined colonization success as well as inland dispersal and speciation mechanisms. We further identified the source areas of the Micronesian biota and reconstructed historical dispersal scenarios: a dominant Austro-Melanesian dispersal scenario, an Indo-Malaysian connecting to the Austro-Melanesian dispersal scenario, and a Neotropical American and an African dispersal scenario toward Micronesia. Most generic origins were estimated between the Eocene and the Miocene and dispersed to Micronesia between the Miocene and the Pleistocene.

Breakdown of self-incompatibility increases opportunities for both self-fertilization and interspecific hybridization, although the latter is dependent on the extent of competition between heterospecific and conspecific pollen. We investigate the mating system and pollination biology of five phylogenetically closely related species within a distylous species complex in Hedyotis L. (Rubiaceae) in southern China. The complex comprises Hedyotis acutangula Champ. ex Benth., Hedyotis shiuyingiae T.Chen, Hedyotis vachellii (Hook. & Arn.) Kuntze, and two putative hybrid species, Hedyotis bodinieri (H.Lév.) Chun and Hedyotis loganioides Benth., hypothesized to result from interbreeding between these species. We test the hypothesis that the breakdown of self- and interspecific incompatibilities in sympatric Hedyotis species might allow interspecific hybridization in natural populations. We assessed the extent of self- and interspecific incompatibility in sympatric populations, including investigations of spontaneous and artificial self-pollination, geitonogamy, inter- and intramorph xenogamy. Artificial interspecific crosses were undertaken between H. acutangula, H. shiuyingiae, and H. bodinieri, between H. acutangula and H. vachellii, and between H. acutangula and H. loganioides. Hedyotis acutangula is demonstrated to be self- and interspecific compatible, whereas H. vachellii, H. bodinieri, and H. loganioides are self-compatible and interspecific incompatible; H. shiuyingiae, in contrast, is strictly self- and interspecific incompatible. Comparisons of pollen tube growth rates in hybridizing species-pairs revealed that heterospecific pollen of H. shiuyingiae, H. vachellii, and H. bodinieri can compete with conspecific self-pollen of H. acutangula. Our study therefore indicates that the breakdown of self-incompatibility directly and indirectly facilitates interspecific hybridization and provides a platform for better understanding evolutionary directionality in Hedyotis.
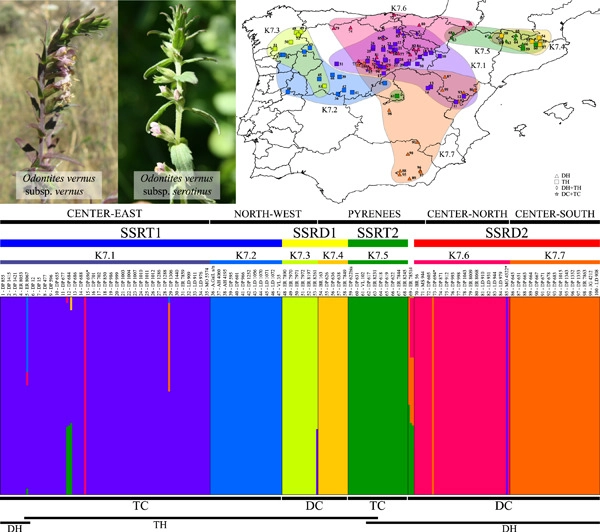
The Odontites vernus group is the most widespread of the genus Odontites, occupying the temperate regions of Eurasia and northern Morocco. The group contains three species, all inhabiting the Iberian Peninsula, where O. vernus s.l. (sensu lato) exhibits remarkable morphological variability and includes diploid and tetraploid individuals corresponding to the two subspecies that occur there. We collected 301 individuals from 100 sampling sites covering the entire distribution of O. vernus in the Iberian Peninsula and genotyped them using 12 simple sequence repeat (SSR) markers. Their ploidy level was estimated by flow cytometry, and two cpDNA regions (rps16 intron and trnK-rps16) were sequenced. We found 129 diploids and 172 tetraploids distributed following a mosaic parapatry model, while only two mixed-ploidy populations were discovered. The 20 haplotypes found fit two well-defined haplogroups, to some extent correlated with estimated ploidy levels. The frequencies of the SSR alleles shared by both cytotypes, as well as those of the private alleles corresponding to the tetraploid cytotype, indicate that tetraploids likely originated at least twice through autopolyploidy. Additionally, the results from SSR markers were structured in a higher number of groups than did the cpDNA sequences. Thus, the genetic distance analysis detected four groups, but the Bayesian analysis of population structure identified seven, with only low levels of gene flow detected among groups. The distributions of the seven genetic groups coincide with well-known refugium areas within the Iberian Peninsula during the climatic oscillations of the Quaternary. Thus, the results give additional support to the “refugia within refugia” hypothesis.
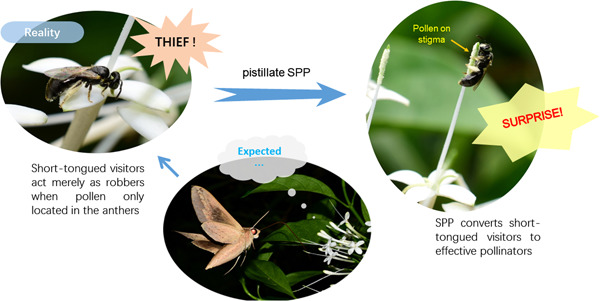
Pollination precision and efficiency have been deemed to be important driving forces in floral evolution. Herkogamy reduction is a main mechanism to increase pollination precision. Secondary pollen presentation (SPP), by which pollen is presented on other floral organs especially pistils, has been widely accepted as a special mechanism to increase pollen transfer precision through spatial reduction of the anther–stigma distance, that is, minimized herkogamy. This overlooks a potential driving force, that is expanding the pollination niche through converting pollen thieves and nectar robbers into effective pollinators. We selected two species as study models with typical pistillate SPP, Pavetta hongkongensis Bremek. (Rubiaceae) and Scaevola taccada (Gaertn.) Roxb. (Goodeniaceae). In both species, two distinct pollinator functional groups were recognized. Short-tongued bees and flies fed on pollen on stigmas but also stole pollen from anthers and robbed nectar, whereas long-tongued hawkmoths and butterflies only collected nectar. Emasculation had no influence on long-tongued pollinators, but significantly decreased the visitation frequency of short-tongued visitors and fruit set, compared to intact flowers, demonstrating short-tongued visitors did not effectively pollinate and acted merely as pollen thieves or nectar robbers when SPP was absent. Data from the two plant species clearly indicated pistillate SPP has additional adaptive advantages of converting ineffective visitors into pollinators and consequently widening the pollination niche, which could help plants overcome environmental stochasticity. Our results suggest that multiple selective forces drive the evolution of SPP and the minimization of herkogamy.
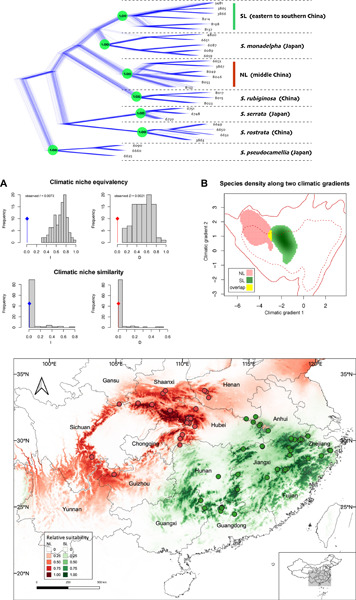
Accurate species delimitation is the key to precise estimation of species diversity and is fundamental to most branches of biology. Unclear species boundaries within species complexes could lead to the underestimation of species diversity. However, species delimitation of species complexes remains challenging due to the continuum of phenotypic variations. To robustly examine species boundaries within a species complex, integrative approaches in phylogeny, ecology, and morphology were applied to the Stewartia sinensis complex (Theaceae) endemic to China. Multispecies coalescent-based species delimitation using 572 nuclear ortholog sequences (anchored enrichment) supported reciprocal phylogenetic monophyly of the northern lineage (NL) and southern lineage (SL), which were not sister clades. Niche equivalency and similarity tests demonstrated significant climatic niche differentiation between NL and SL with observed Warren et al.'s I = 0.0073 and Schoener's D = 0.0021. Species distribution modeling also separated their potential distribution. Morphometric analyses suggested significant interlineage differentiation of multiple traits including the ratio of length and width, leaf width, and pedicel length, although overall similarity did not differ. Based on the integrative species concept, two distinct species were proposed with legitimate names of Stewartia gemmata for SL and S. sinensis for NL. Our empirical study of the S. sinensis complex highlights the importance of applying multiple species criteria, in particular the underappreciated niche differentiation, to species delimitation in species complexes pervasive in plants.
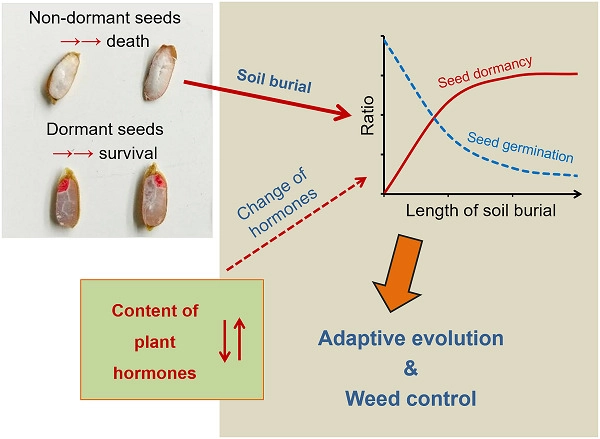
Seed dormancy plays a key role in preventing seeds of higher plants from random germination under adverse environmental conditions. Previous studies suggested that a critical temperature could regulate germination of weedy rice seeds without primary dormancy at seed dispersion. However, what will happen to the non-dormant seeds after shattering in the soil seed banks when temperature fluctuates to exceed the critical temperature remains an interesting question to be investigated. To determine whether or not soil burial can change the status of dormancy in weedy rice seeds, we examined germination ratios of weedy rice seeds after soil-burial treatments. In addition, we compared hormone levels in the untreated seeds and viable but ungerminated seeds after soil burial. Results showed that soil burial induced a proportion of 41%–72% dormant seeds in the initially non-dormant weedy rice seeds. Also, the induction of seed dormancy is associated with the change of hormone levels in the seeds treated by soil burial, suggesting that soil burial can significantly activate the control of hormone production in seeds. Together, the previously reported mechanism of critical temperature-inhibited seed germination and the newly found phenomenon of soil burial-induced seed dormancy provide a “double-security” strategy to ensure germination of weedy rice seeds under a favorable condition in agricultural ecosystems. The findings not only reveal the important role of rapid evolution of adaptive functions in weeds, such as weedy rice, in adapting to changing agricultural environments, but also facilitate the design of strategies for effective weedy rice control practices.
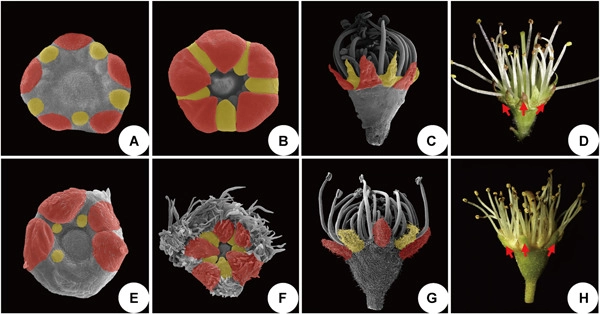
Although the vast majority of Prunus L. (Rosaceae) species have clearly differentiated sepals and petals, two former genera Maddenia and Pygeum have been described as having an undifferentiated perianth. However, floral morphological and morphogenetic data are scarce, and a renewed investigation is essential to understand the evolution of the perianth differentiation. Here, floral morphogenesis in Prunus hypoleuca (Koehne) J.Wen (=Maddenia hypoleuca Koehne) and Prunus topengii (Merr.) J. Wen & L. Zhao (=Pygeum topengii Merr.) were examined with scanning electron microscopy. The floral development demonstrates that the ten perianth parts can be distinguished as five sepals in an external whorl and five petals in an internal whorl. The sepal primordia are broad, crescent-shaped, and truncate. The petal primordia are rounded and initially resemble the androecium. However, at maturity petals and sepals look much the same in the two species, differing from other Prunus species. The ovule is anatropous and unitegmic, but there is a basal appendage near the ovule of P. hypoleuca which is absent in P. topengii. The direction of development of floral nectaries in the hypanthium is basipetal in P. hypoleuca but acropetal in P. topengii. Perianth segments are differentiated in the two groups and the similarity of the perianth parts is secondarily acquired. Our results support the separation of the Maddenia and Pygeum groups as well as their inclusion in a broader monophyletic Prunus based on molecular phylogenetic studies. We herein provide a new nomenclatural change: Prunus topengii (Merr.) J. Wen & L. Zhao, comb. nov.

Sexual dimorphism in dioecious plants often occurs as a consequence of the different resource requirements of females and males, especially during reproduction. The contrasting reproductive roles of the sexes can influence the phenology of growth, plant size, and flowering time, with implications for the intensity of competitive interactions within and between the sexes. Here, we investigate the influence of contrasting nutrient regimes and intra-sexual and inter-sexual competition on the expression of sexual dimorphism in life-history traits and biomass allocation throughout the life cycle of the dioecious annual Rumex hastatulus Baldw. (Polygonaceae). Development of a sex-specific marker enabled us to quantify the influence of competition on sex-specific differences in mortality and vegetative traits. We were particularly interested in determining whether the overall performance of the sexes might differ between the two forms of intra-specific competition, potentially providing evidence for sexual specialization in resource acquisition and niche differentiation. Our results indicated that although patterns of sexual dimorphism were dynamic, they were largely insensitive to nutrient conditions. We found that intra-sexual competition was more severe than inter-sexual competition, differentially affecting mortality and most traits during the vegetative and particularly the reproductive stage of the life history. Female trait values generally increased more under inter-sexual than intra-sexual competition in comparison to males. Our findings are consistent with temporal niche differentiation resulting from sexual specialization for different resource requirements and provide evidence for the “Jack Sprat effect.”
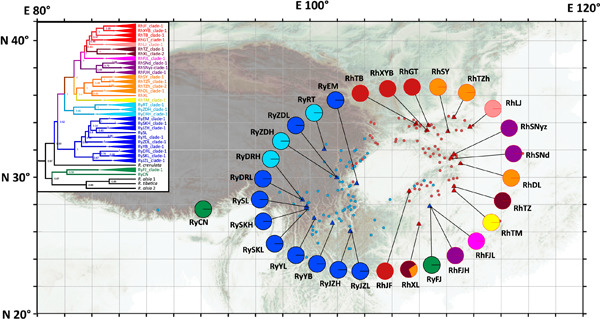
A ring species consists of two reproductively isolated forms connected by a chain of intergrading populations encircling a geographic barrier. The mountains encircling the Sichuan Basin in southwestern China harbor great species diversity and endemism, and they are candidate regions for ring species. Here, we examined a potential ring species complex with a ring distribution surrounding the Sichuan Basin and the reproductive barrier between sibling species Rhodiola yunnanensis and R. henryi of the R. yunnanensis complex. In this study, we test the hypothesis that R. yunnanensis and R. henryi diverged by the ring-species model using an amplicon sequencing strategy targeting the introns of 27 single-copy nuclear genes and 14 chloroplast DNA sequences. Our studies indicated that the R. yunnanensis complex is monophyletic, originates at the late Miocene, and forms its current ring distribution pattern after the LIG. In addition, clear genetic intergradation was not found among R. henryi populations within the distribution ring. All these findings suggest that the divergence of two sibling species was not driven by the geographic isolation, and they were not originated from the ring-species model; however, the basin-surrounding distribution pattern and reproductive barrier between them meet some criteria for being a ring species.

The Hengduan Mountains region (HMR) on the southeastern Tibetan Plateau, supports a high diversity of herbs, particularly in its subalpine to alpine ecosystems, due to high altitude and cool temperate climate. Current understanding on the formation of such herbaceous richness is based chiefly on molecular phylogenies; however, direct geological evidence is lacking because herbs are rarely preserved as macroscopic fossils. In this study, we present abundant fossil fruits and seeds of herbs from the late Pliocene Heqing Basin in the southern HMR. Our systematic analysis shows the presence of at least 18 species belonging to 11 genera, that is, Ranunculus, Corydalis, Rumex, Polygonum, Chenopodium, Stellaria, Fragaria, Astragalus, Aster, Carex, and Schoenoplectus, of which Polygonum is most abundant followed by Astragalus. This finding throws the first light from fossil evidence on the rise of herbaceous diversity in the region. We interpret the local assembly of these herbs as resulting from rapid pre-Pliocene species diversifications of many herbaceous groups in the HMR. As nowadays most of these herbs grow primarily in meadows and a few occur as subaquatic plants, we suggest an open meadow hosting some scattered shrubs in the vicinity of a vegetated wetland in the Heqing Basin during the late Pliocene. This provides the first direct evidence of past treeless open vegetation within the HMR and thus improves our knowledge of vegetation evolution in the region. We suggest that the uplift-induced climate cooling and monsoon-associated precipitation seasonality are potentially the key driving forces for the opening of meadow vegetation in the HMR.
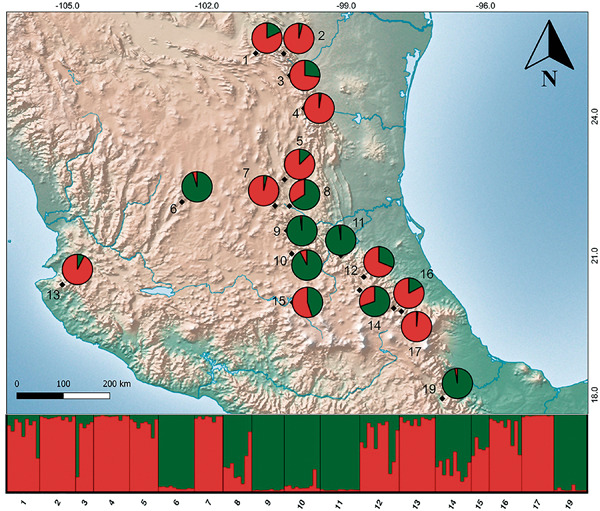
Differentiation among populations, sometimes despite ongoing gene exchange, is a key step in speciation. Therefore, comparison of intra- and interspecific differentiation patterns is of great significance to understanding speciation. The genus Quercus is an interesting system to test speciation models in the presence of gene flow, due to its weak interspecific reproductive barriers. The aim of the present study was to characterize the degree and pattern of morphological and genetic differentiation among different morphotypes in the white oak Quercus laeta, some corresponding to the previously described species Quercus centralis, Q. laeta, Quercus prinopsis, and Quercus transmontana, as well as geographically structured variation within Q. transmontana not described previously. Our goal was to evaluate if some of these can be considered distinct specific entities or are rather part of a continuum of variation. Nine microsatellite loci and two intergenic regions of chloroplast DNA were analyzed. Morphological differences were evaluated using geometric morphometrics. Chloroplast DNA showed low differentiation, suggesting introgression or sharing of ancestral haplotypes among the Q. laeta morphotypes. Nuclear microsatellites indicated differentiation into two distinct main genetic groups, which were congruent with morphological differentiation. In conclusion, nuclear markers and morphological variations suggest the existence of at least two different entities within Q. laeta.
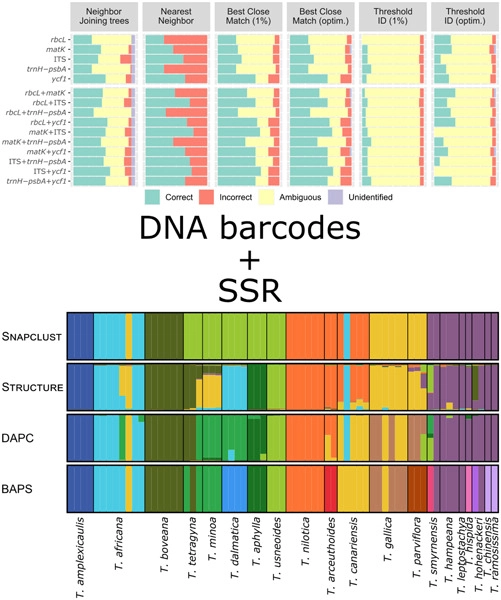
DNA barcoding allows the identification of an organism by comparing the sequence of selected DNA regions (barcodes) with a previously compiled database, and it can be useful for taxonomic identification of species in complex genera, such as Tamarix. Many species of this genus show convergent morphology, which leads to frequent errors in their identification. Highly variable genetic markers, such as microsatellites or short sequence repeats (SSR), could be used to differentiate species where DNA barcodes fail. Here, we tested the ability of both, 5 different marker regions (rbcL, matK, ITS, trnH-psbA, and ycf1), and 14 microsatellites, to properly identify Tamarix species, especially those from the Mediterranean Basin, and compared the pros and cons of the different analytical methods for species identification. DNA barcoding allows the genetic identification of certain species in Tamarix. The two-locus barcodes matK + ITS and ITS + ycf1 were the best-performing combinations, allowing up to 69% and 70%, respectively, correct identification. However, DNA barcoding failed in phylogenetically close groups, such as many Mediterranean species. The use of SSR can aid the identification of species, and the combination of both types of data (DNA barcoding and SSR) improved the success. The combination of data was especially relevant in detecting the presence of hybridization processes, which are common in the genus. However, caution must be exercised when choosing the clustering methods for the SSR data since different methods can lead to very different results.
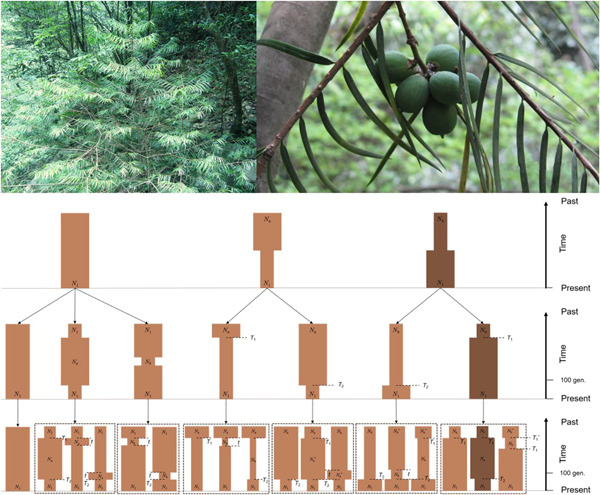
Both paleoclimatic change and anthropogenic habitat destruction can have adverse effects on species demography and, in turn, could lead a species towards being endangered and rare. Understanding the relative importance of these natural and anthropogenic factors driving species endangerment and rarity is thus crucial for effective conservation planning but remains elusive. Here, we examine the phylogeography and demographic history of an endangered conifer species in China, Torreya jackii Chun, and assess the relative importance of natural and anthropogenic factors that might have put the species in its endangered state. We collected tissue samples from all the 13 extant wild populations, and analyzed the genetic variation using eight nuclear microsatellites and four chloroplast and one mitochondrial DNA fragments. We found low genetic and nucleotide diversities, which could explain the absence of spatial and phylogeographic structure. Using a hierarchical approximate Bayesian computation technique, we identified the demographic scenario that best fits the genetic data and found that effective population size was low at least 200 000 years ago but expanded after the last glacial maximum (LGM). The paleoclimatic niche model revealed a profound effect of precipitation on the distribution of T. jackii and predicted that the current distribution areas were suitable during the LGM. Despite the post-LGM expansion, the best-supported scenario showed a dramatic population collapse during the past 300 years, when anthropogenic disturbances also increased dramatically. Overall, our study sheds light on how historical factors and human impacts jointly threaten the persistence of a species, and these aspects should be duly considered in species conservation planning.

We describe six specimens consisting of cranial remains and associated partial presacral axial series belonging to ornithuromorph birds from the Changma locality of the Lower Cretacous Xiagou Formation of northwestern Gansu Province, China. Comparison among specimens is limited by the paucity of overlapping elements, their differing exposed views, and, in some specimens, poor preservation. Despite this, three separate taxa are represented, evidenced by differences in their dentary dentitions: one specimen is edentulous, another has sharp, closely spaced, relatively high-crowned and peg-like teeth, and a third preserves blunt, relatively low-crowned teeth placed in a communal groove, a morphology previously reported among adult birds only in Hesperornithiformes. We propose that the high-crowned specimen may be referred to Gansus yumenensis based on shared similarities with the closely related Iteravis huchzermeyeri, including a very similar dentition and an edentulous premaxilla with elongate, unfused frontal processes and no palatal processes. The two other specimens are considered new taxa, for which we erect the names Meemannavis ductrix gen. et sp. nov. and Brevidentavis zhangi gen. et sp. nov. These new specimens confirm that the Changma locality is dominated by ornithuromorph birds and contribute to a better understanding of this important avifauna. The observed variation in dental morphology hints at trophic diversity like that observed in ornithuromorphs from the penecontemporaneous Jehol Group of northeastern China.
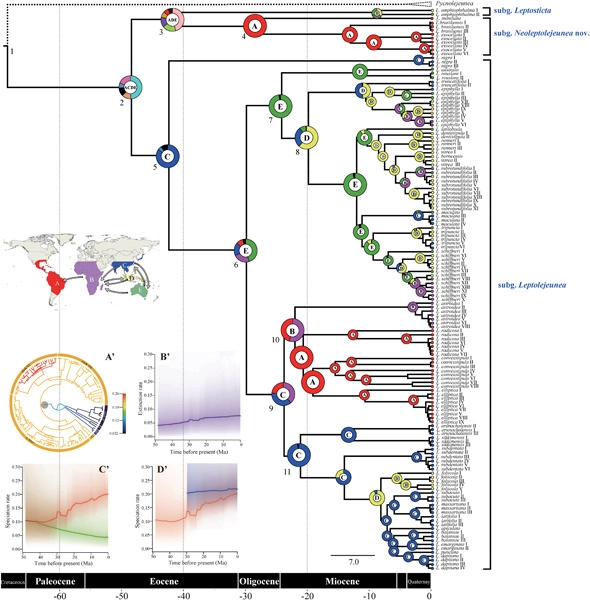
Although recent molecular phylogenetic analyses have greatly improved our understanding of the classification of the large liverwort family Lejeuneaceae, the frequent incongruencies between morphology-based taxonomy and molecular phylogeny have hindered our understanding of evolutionary diversification within the group. Here we focus on Leptolejeunea (Spruce) Steph., a pantropical epiphyllous genus in Lejeuneaceae with 40 species. Phylogenetic studies on the genus have been hampered by insufficient taxon sampling, leaving the deep phylogenetic relationships within this group unresolved. We present the most comprehensive phylogenetic analysis of the genus to date with sampling of over 80% of species, including the enigmatic Leptolejeunea spinistipula (Mizut.) X.L.He endemic to Borneo. Based on data from three molecular markers with representatives of Leptolejeunea and its allies, Leptolejeunea appeared to be monophyletic following the exclusion of L. spinistipula and its transfer to Soella R.L.Zhu, L.Shu, Qiong He & Y.M.Wei. A total-evidence approach was taken to resolve the backbone phylogeny of Leptolejeunea and a first infrageneric classification of Leptolejeunea, including a new subgenus and three new sections, is proposed based on integrated molecular and morphological evidence. Reconstruction of the evolutionary history showed a wide ancestral area of Leptolejeunea during the Paleogene that arose in mainland Asia, followed by an accelerated speciation rate. Across the biogeographical history of Leptolejeunea, long-distance dispersal had profound effects on population expansion. Our findings suggest that Australasia is a source of biodiversity of Asian evergreen broad-leaved forests that have been established since the Oligocene and rose after the early Miocene.
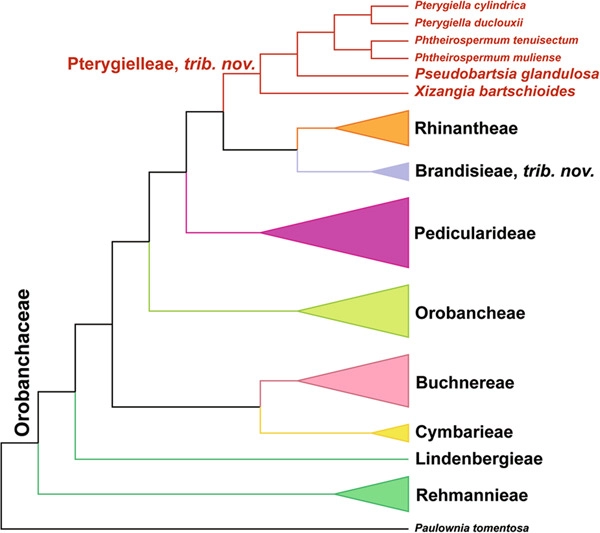
The millions of herbarium specimens in collections around the world provide historical resources for phylogenomics and evolutionary studies. Many rare and endangered species exist only as historical specimens. Here, we report a case study of the monotypic Pseudobartsia yunnanensis D. Y. Hong (=Pseudobartsia glandulosa[Bentham] W. B. Yu & D. Z. Li: Orobanchaceae) known from a single Chinese collection taken in 1940. We obtained genomic data of Pseudobartsia glandulosa using high-throughput short-read sequencing, and then assembled a complete chloroplast genome and nuclear ribosome DNA region in this study. We found that the newly assembled three plastid DNA regions (atpB-rbcL, rpl16, and trnS-G) and nuclear ribosomal internal transcribed spacer (nrITS) of Pseudobartsia glandulosa were more than 99.98% similar to published sequences obtained by target sequencing. Phylogenies of Orobanchaceae using 30 plastomes (including 10 new plastomes), using both supermatrix and multispecies coalescent approaches following a novel plastid phylogenomic workflow, recovered seven recognized tribes and two unranked groups, both of which were proposed as new tribes, that is, Brandisieae and Pterygielleae. Within Pterygielleae, all analyses strongly supported Xizangia D. Y. Hong as the first diverging genus, with Pseudobartsia D. Y. Hong as sister to Pterygiella Oliver + Phtheirospermum Bunge (excluding Phtheirospermum japonicum [Thunberg] Kanitz); this supports reinstatement of Pseudobartsia and Xizangia. Although elements of Buchnereae-Cymbarieae-Orobancheae and Brandisieae-Pterygielleae-Rhinantheae showed incongruence among gene trees, the topology of the supermatrix tree was congruent with the majority of gene trees and functional-group trees. Therefore, most plastid genes are evolving as a linkage group, allowing the supermatrix tree approach to yield internally consistent phylogenies for Orobanchaceae.












