



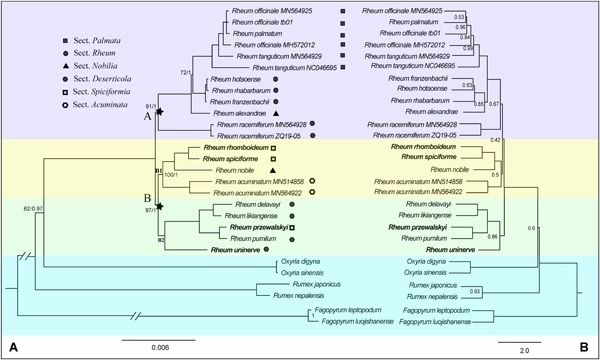
Species of Rheum have high medicinal value, with the center of diversity in the Qinghai–Tibet Plateau (QTP) and adjacent regions. However, phylogenetic relationships of Rheum are still unclear due to fragment markers providing insufficient informative loci. Here, we sequenced and annotated plastomes of nine Rheum species, and compared the genome structure among the novel nine species along with three published species. Comparative analyses revealed that plastomes of Rheum share a relatively conserved structure. Five highly divergent regions (accD, ccsA, matK, ndhF, and ndhH) can be used as valuable molecular markers for further species delimitation and population genetic studies. Twenty-two accessions representing 17 species were used for phylogenetic analysis, which generated a robust phylogenetic tree and revealed two major clades within Rheum. Phylogenetic results showed that glasshouse structures and cushions of Rheum are results of parallel evolution during adaptation to similar environments. Inconsistent tree topology between concatenated and coalescent methods was detected, implying that incomplete lineage sorting and hybridization may have occurred in the evolutionary history of Rheum. Divergence time estimation based on two fossil calibrations and three secondary calibrations revealed a Miocene to middle Oligocene origin of Rheum. Our study provides valuable genomic resources for the medicinally important genus Rheum, while gaining helpful insights into its systematics and evolution.
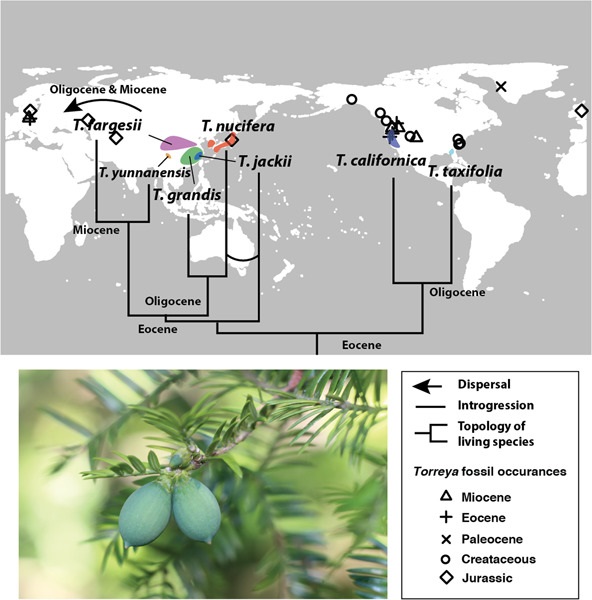
Restriction site-associated DNA sequencing (RAD-seq) enables obtaining thousands of genetic markers for phylogenomic studies. However, RAD-seq data are subject to allele dropout (ADO) due to polymorphisms at enzyme cutting sites. We developed a new pipeline, RAD-seq Allele Dropout Remedy in our study of the gymnosperm genus, Torreya, to mitigate ADO in outgroups by recovering missing loci from previously published transcriptomes. By using RADADOR to supplement Rad-seq data in combination with plastome and mitochondrial gene sequences, morphology, and fossil records, we reconstructed the phylogenetic and biogeographic histories of the genus and tested hypotheses on anomalies of biodiversity of the eastern Asian-North American floristic disjunction. Our results showed that our pipeline recovered many loci missing from the outgroup, and the improved data yielded a more robust phylogeny for Torreya. Using the fossilized birth–death model and divergence–extinction–cladogenesis method, we resolved a detailed biogeographic history of Torreya that suggested a Jurassic origin spanning Laurasia and differential speciation and extinction among continents accounting for modern diversity, which is biased toward eastern Asia (EA). The biogeographic results also supported a vicariance origin of modern Torreya from a widespread ancestor in EA and North America (NA) in the mid-Eocene, and cross Beringian exchange in the early Paleogene before the vicariant isolation, in contrast to the “out of NA” pattern common to gymnosperms and to the “out of EA” hypothesis previously proposed for the genus. Furthermore, we observed phylogenetic discordance between the nuclear and plastid phylogenies for Torreya jackii, suggesting differential lineage sorting of plastid genomes among species of Torreya or plastid genome capture in T. jackii.

The olive genus Olea includes c. 30–40 taxa in three subgenera (Olea, Tetrapilus, and Paniculatae) within the family Oleaceae. Historically, the Olea genus was classified into four groups that were overall well supported by reconstructed phylogenies, despite incomplete sampling of subgenus Tetrapilus and poor resolution within clades. These analyses also showed that the genus was not monophyletic. Reliable identification of Olea species is important for both their conservation and utilization of this economically important genus. In this study, we used phylogenomic data from genome skimming to resolve relationships within Olea and to identify molecular markers for species identification. We assembled the complete plastomes, and nrDNA of 26 individuals representing 13 species using next-generation sequencing and added 18 publicly available accessions of Olea. We also developed nuclear SNPs using the genome skimming data to infer the phylogenetic relationships of Olea. Large-scale phylogenomic analyses of 138 samples of tribe Oleeae supported the polyphyly of Olea, with Olea caudatilimba and Olea subgenus Tetrapilus not sharing their most recent common ancestor with the main Olea clade (subgenus Paniculatae and subgenus Olea). The interspecific phylogenetic resolution was poor owing to a possible rapid radiation. By comparing with the plastome data, we identified the markers ycf1b and psbE-petL as the best Olea-specific chloroplast DNA barcodes. Compared with universal barcodes, specific DNA barcodes and super-barcode exhibited higher discriminatory power. Our results demonstrated the power of phylogenomics to improve phylogenetic relationships of intricate groups and provided new insights into barcodes that allow for accurate identification of Olea species.

Understanding hybridization and introgression between natural plant populations can give important insights into the origins of cultivated species. Recent studies suggest differences in ploidy might not create such strong reproductive barriers as once thought, and thus studies into cultivated origins should examine all co-occurring taxa, including those with contrasting ploidy levels. Here, we characterized hybridization between Chrysanthemum indicum L., Chrysanthemum vestitum (Hemsley) Ling and Chrysanthemum vestitum var. latifolium (Zhou & Chen), the most important wild species involved in the origins of cultivated chrysanthemums. We analyzed the population structure of 317 Chrysanthemum accessions based on 13 microsatellite markers and sequenced chloroplast trnL-trnF for a subset of 103 Chrysanthemum accessions. We identified three distinct genetic clusters, corresponding to the three taxa. We detected 20 hybrids between species of different ploidy levels, of which 19 were between C. indicum (4x) and C. vestitum (6x) and one was between C. indicum and C. vestitum var. latifolium (6x). Fourteen hybrids between C. indicum and C. vestitum were from one of the five study sites. Chrysanthemum vestitum and C. vestitum var. latifolium share only one chloroplast haplotype. The substantially different number of hybrids between hybridizing species was likely due to different levels of reproductive isolation coupled with environmental selection against hybrids. In addition, human activities could play a role in the different patterns of hybridization among populations.
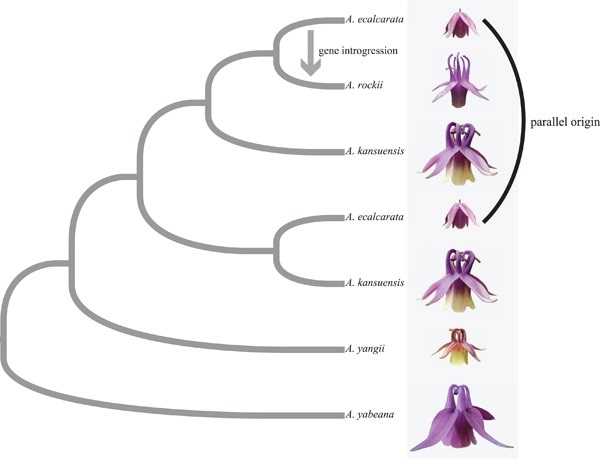
Traits are basic attributes of organisms that form the basis for speciation and diversity. The floral nectar spur is a classic example of a key innovative trait. Differences in nectar spur morphology can lead to pollinator specialization and thereby promote reproductive isolation between species. Despite its importance, the nectar spur has been lost in some members of the columbine genus (Aquilegia), resulting in a new spurless trait, and the evolutionary influence of this trait has become a topic of scientific interest. Aquilegia ecalcarata is an important representative columbine species that lacks spurs. Here, we resequenced the genomes of 324 individuals from A. ecalcarata and four related species. We found that A. ecalcarata was divided into three groups based on the phylogenetic relationships and population genetic structures. Topology weighting analysis revealed that A. ecalcarata has multiple origins, and Patterson′s D statistic showed that the spurless trait may have one origin. Floral morphological analysis revealed significant differences between A. ecalcarata and its spurred sister groups, and the floral phenotypes of the three A. ecalcarata groups have identical or similar floral phenotypes. Our results confirmed that the spurless trait not only produced the phenotype of A. ecalcarata but also contributed to the emergence of the A. rockii phenotype. Moreover, the spurless trait promoted the divergence between A. ecalcarata and its close, spurred relatives. Our research shows that the loss of key innovative traits can play a very important role in speciation and species diversity.
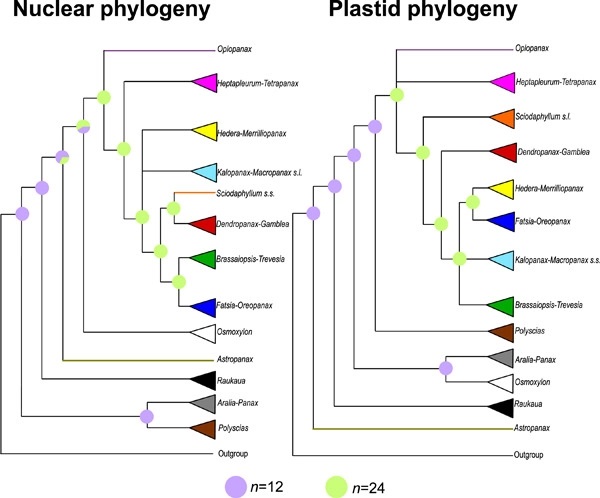
The phenomenal advances in sequencing techniques and analytical development during the last decade have provided a unique opportunity to unravel the evolutionary history of lineages under complex patterns of evolution. This is the case of the largest clade of the ginseng family (Araliaceae), the Asian Palmate group (AsPG), where the large internal polytomies and genome incongruences detected in previous studies pointed to a scenario of radiation with hybridization events between genera for the early evolution of the group. In this study, we aim to obtain well-resolved nuclear and plastid phylogenies of the AsPG using Hyb-Seq to evaluate the radiation hypothesis and assess the role of hybridization in the early evolution of the group. We performed concatenated- and coalescent-based phylogenetic analyses from the 936 targeted nuclear loci and 261 plastid loci obtained for 72 species representing 20 genera of the AsPG and the main clades of Araliaceae. The impact of hybridization and incomplete lineage sorting (ILS) was assessed with SNaQ, and genome duplications were evaluated with ChromEvol. Our nuclear and plastid phylogenies are compatible with a scenario of early radiation in the AsPG. Also, the identification of extensive signals of hybridization and ILS behind the genome incongruences supports hybridization as a major driving force during the early radiation. We hypothesize a whole-genome duplication event at the origin of the AsPG, followed by a radiation that led to extensive ILS, which, alongside the early inter-genera hybridization, is obscuring the phylogenetic signal in the early evolution of this major clade.
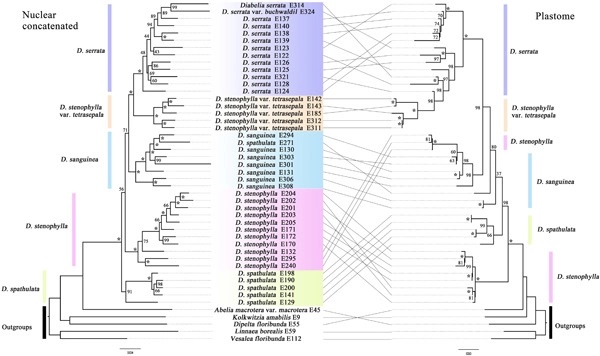
Understanding biological diversity and the mechanisms of the Sino-Japanese disjunctions are major challenges in eastern Asia biogeography. The Sino-Japanese flora has been broadly studied as an ideal model for plant phylogeography. Diabelia Landrein (Caprifoliaceae) is an East Asian genus, with a disjunctive distribution across the Sino-Japanese region. However, relationships within Diabelia remain elusive. In this study, we reconstructed the phylogeny of Diabelia and inferred historical biogeography and evolutionary patterns based on nuclear and plastid sequences from target enrichment and genome skimming approaches, respectively. We found that the main clades within Diabelia were discordant between nuclear and plastid trees. Both nuclear and plastid phylogenetic analyses supported five main clades: Diabelia serrata (Siebold & Zucc.) Landrein, Diabelia tetrasepala (Koidz.) Landrein, Diabelia sanguinea (Makino) Landrein, Diabelia stenophylla (Honda) Landrein, and Diabelia spathulata (Siebold & Zucc.) Landrein. Species network analyses revealed that Diabelia tetrasepala is likely the result of a hybridization event. Divergence time estimation and ancestral area reconstructions showed that Diabelia originated in Japan during the early Miocene, with subsequent vicariance and dispersal events between Japan and Korea, and between Japan and China. Overall, our results support the division of Diabelia into five main clades and the recognition of five species in the genus. This research provides new insights into the species delimitation and speciation processes of taxonomically complex lineages such as Diabelia.
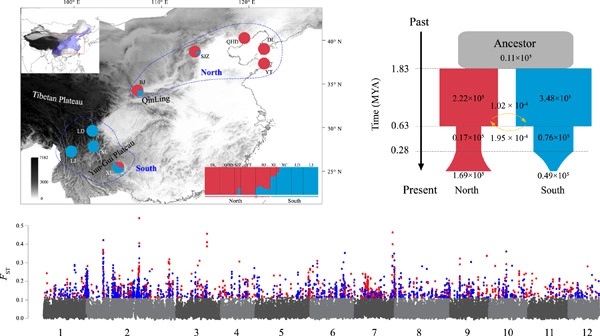
Multiple evolutionary forces contribute to heterogeneous genomic landscapes; however, disentangling their relative contributions is challenging. We sampled nine populations across the distribution of Quercus dentata, a dominant forest tree in East Asia, and used whole-genome sequencing data to investigate mechanisms underlying divergence. We identified two genetic groups (north and south) that diverged ~1.84 million years ago, consistent with the uplift of the Qinling Mountains during the Pleistocene. The north group experienced a bottleneck during the middle–late Pleistocene and expanded from multiple refugia. The south group experienced a more severe bottleneck and showed high population differentiation, probably due to long-term isolation and habitat fragmentation. We detected genomic islands with elevated relative differentiation (FST) scattered across the genome. Among these, 65.9% showed reduced absolute divergence (dXY) consistent with linked selection, while the remaining (34.1%) showed elevated dXY suggestive of divergent sorting of ancient polymorphisms. The recombination rate in genomic islands was lower than background, suggesting the importance of genome structure in shaping the genomic landscape. We detected 108 single nucleotide polymorphisms significantly associated with environmental factors, 12 of which clustered in a region of ~500 kb. This region showed multiple signals of positive selection in the north group, including the enrichment of XP-extended haplotype homozygosity scores, an elevated population branch statistic, and an excess of high-frequency derived alleles. In addition, we found that linkage disequilibrium was low and derived haplotypes declined rapidly in this region, indicating selection on standing variation. Our results clarify the evolutionary processes driving genomic divergence in Q. dentata.

The southwest mountainous region of China has been characterized as one of the worldwide biodiversity hotspots, but mechanisms underlying diversification of organisms in this region are still not clear. We assessed whether fragmented mountainous habitats and Pleistocene climate changes impacted the genetic diversity and diversification patterns of the hoary bamboo rat (Rhizomys pruinosus Blyth), a widely distributed species of rodent in SW China. Genetic diversity analyses were undertaken based on four mitochondrial DNA regions and 12 nuclear microsatellite loci (simple sequence repeats), representing 153 individuals from 24 populations across SW China. Moreover, we investigated correlations between genotype and geographical components, and predicted species distribution models for R. pruinosus under the historical and present climate conditions. Both mitochondrial DNA and simple sequence repeat markers revealed substantial genetic diversity and strong differentiation between populations. Phylogeographical analyses revealed two phylogenetic clades that were consistent with their geographical distributions (eastern and western clades). We inferred that the divergence of R. pruinosus was largely driven by Quaternary climatic oscillations and regionally fragmented mountainous habitats with environmental and geographical heterogeneity. Overall, our study revealed diversification patterns of R. pruinosus—patterns that may be shared by small alpine vertebrates in SW China.

Nectar, the most common floral reward, is generally used to determine whether an orchid species involves deceptive pollination. Estimates of the deceptive pollination systems with nectarless flowers have ranged from one quarter to one third of the nearly 30 000 species of orchids. These estimates, however, are biased towards temperate-zone, usually terrestrial, orchids. Here we investigated nectar production and property in 34 epiphytic orchid species of the Southeast Asian genus Dendrobium. Twenty-one species were observed producing nectar. The amount and sugar concentration (in bagged flowers) of 12 species varied from 0.45 to 2.78 μL and from 8.1% to 31.1%. The nectar was sucrose-dominant, typical of bee-pollinated flowers. Reconstruction of phylogenetic relationship indicated that transition of nectar secretion occurred in the genus. Spur length was positively correlated with flower size but species with relatively long spurs tended to produce small volume of nectar. Nectar production was strikingly variable among and within individuals in some species, suggesting that a vital measurement of bagged and fresh flowers is needed. Given that the quantitative measurement of nectar or floral reward in orchid species remains scarce, an estimate of deceptive pollination systems awaits further survey in diverse genera.

Triterpene scaffolds are cyclized by differential functional genes of the oxidosqualene cyclase (OSC) gene family, which are widely present in plant species. Thirty-nine OSCs were identified from the hexaploid bread wheat (Triticum aestivum L.) genome. The gene structure organization and the distribution of conserved motifs of OSCs gene family revealed that the TaOSC proteins are highly conserved. By phylogenetic analysis, all OSC proteins from wheat and other cereal crops were clustered into 11 subfamilies (I–XI). Based on the publicly available transcriptional profiling, the tissue-specific expression patterns of TaOSC during several developmental stages revealed diverged expression patterns, indicating that the TaOSC family has undergone subfunctionalization. Furthermore, homeolog expression bias was commonly detected in the TaOSC subfamilies, such as the expression of Ta5A004900 and Ta5D011800 in the subfamily II were specifically expressed in the leaf sheath and flag leaf sheath. In addition, two genes were strongly induced by Pst, indicating that the two homolog genes are involved in the resistance to powdery mildew. Molecular evolution analysis indicated that the TaOSC family genes were undergone purifying selection. So, the powdery mildew response may be very conservative and important biological function of the Ta5A004900 and Ta5D011800 in wheat. In conclusion, these results can be used to explore the diversity of triterpenes and clarify the mechanism of triterpene cyclization, and also provide useful information for future studies on the function of the TaOSC gene family in bread wheat and other important crops.
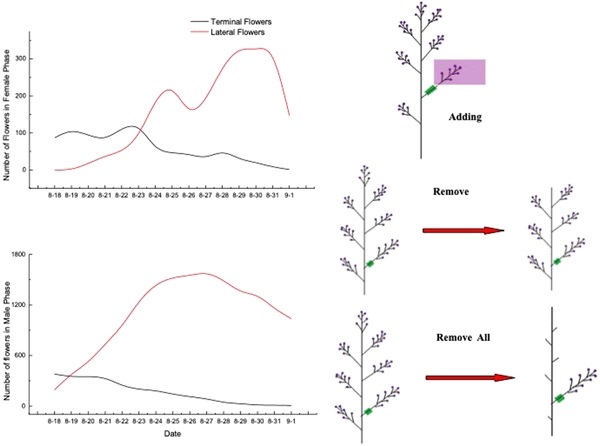
Flowering phenology and clonal growth are known to affect resource and pollen availability, and therefore select for adaptive or constrained sex allocation strategies to some degree. However, the consequences of temporal sex allocation patterns for reproductive fitness across the flower, inflorescence, and genet levels have rarely been examined. Moreover, experimental tests of the underlying regulatory mechanisms are scarce. We examined the association of flowering phenology and inflorescence position with temporal sex allocation and reproductive success in the protandrous perennial clonal herb, Aconitum kusnezoffii, over four consecutive growing seasons by examining more than 39 000 flowers. We also conducted controlled experiments to test the effects of resource and pollen limitation on the female reproductive success of lateral inflorescences. We found that some male functions were positively correlated with flowering phenology, whereas female reproductive success was negatively correlated with flowering phenology and inflorescence position. Lateral inflorescences invested more in male function than terminal inflorescences and therefore yielded fewer and smaller seeds. Resource limitation may serve as the key mechanism underlying this differentiated pattern. Decreased female reproductive success was consistently observed at the flower and inflorescence levels as flowering occurred later in the growth season. Late-blooming lateral inflorescences specialized in the male function, and their female reproductive success was constrained by early-blooming terminal inflorescences. This might be the first attempt to systematically demonstrate sex allocation strategy differentiation in a protandrous plant species at the inflorescence level. In addition, our study provides empirical evidence of dichogamy selecting for specialized sex allocation strategies among inflorescences.
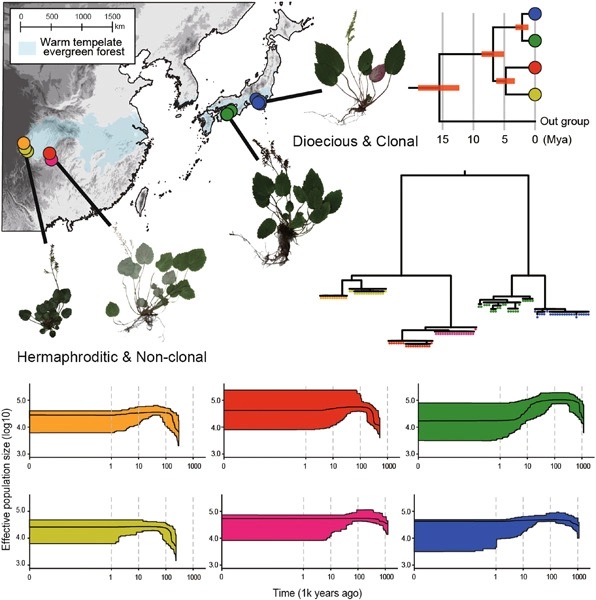
Tertiary relicts often show evolutionary stasis in morphology and ecology and have been hypothesized to retain stable population sizes in refugia. However, recent studies have reported that some relicts evolutionarily shifted their physiology, ecology, and morphology and experienced various patterns of demography. To understand the historical survival of relict plants, a multidimensional study investigating the evolution of ecological and morphological traits as well as population demographic history is needed. The genus Tanakaea (Saxifragaceae) comprises two species in China and Japan. These species share most vegetative characteristics and are sometimes treated as a single species. The distribution pattern is relictual, as the populations are confined to small areas in mesic warm temperate forests less influenced by Quaternary glacial climates. Focusing on the relictual plant group, this study tested the hypotheses of evolutionary stasis and population stability in long-term refugia. Genetic analyses using plastome sequences and genome-wide single-nucleotide polymorphisms revealed divergence of the two species approximately 6.8 million years ago and strong genetic differentiation of the regional populations. Demographic analysis revealed that almost all populations retained stable population sizes during glacial–interglacial climate changes, supporting the traditional view. However, morphological assessments revealed a simultaneous shift in breeding systems (from hermaphrodite to dioecy/non-clonal to clonal reproduction) in Japanese species and intraspecific differentiation of leaf traits. Therefore, the relict species do not show evolutionary stasis in every aspect. Changes in reproductive characteristics may have contributed to their long-term in situ survival.
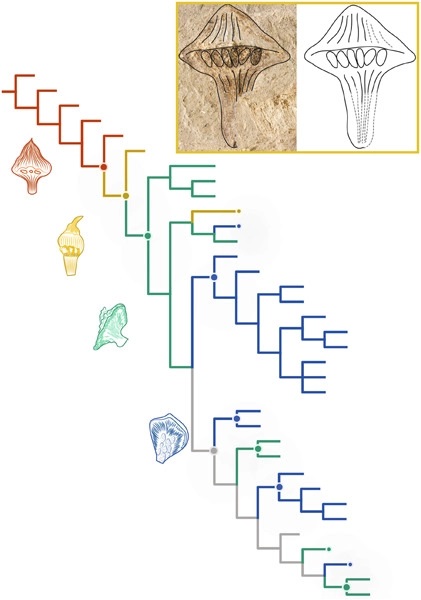
The conifer family Cupressaceae encompasses seven subfamilies. Five of them were once considered to constitute the family Taxodiaceae, later eliminated because of its paraphyletic nature but remaining as an informal category for early-diverging Cupressaceae lineages. Among the taxodiaceous subfamilies, Athrotaxoideae shows a unique morphology in its ovuliferous complexes (OCs) and a phylogenetically unexplored fossil record. We describe the new genus and species Patagotaxodia lefipanensis, based on OC adpressions associated with leafy branches collected at the Maastrichtian section of the Lefipán Formation (Patagonia, Argentina), and we refer it to Athrotaxoideae. We include Patagotaxodia in total evidence phylogenetic analysis to test its affinity, and we recover it within the subfamilies Athrotaxoideae or Cunninghamioideae. However, we argue that the characters supporting the athrotaxoid affinity are more meaningful in a taxodiaceous systematic context. This placement is also supported by taxon inclusion-exclusion experiments. We discuss the position of other Cretaceous athrotaxoid records. With basis on the morphological insights provided by the OC morphology of extant and extinct Athrotaxoideae, we study the evolution of the OC morphology in the family in a phylogenetic context and discuss the results in the light of the fossil record of the family. We discuss how and when the different morphologies appeared in the family. Based on phylogenetic, temporal, morphological, and ontogenetic evidence, we conclude that the OC morphology shown by the subfamily Athrotaxoideae is intermediate between two of the most common morphologies within extant and extinct Cupressaceae species, one of which would show adaptative advantages over basal morphologies.

The fossil record evidences an old origin and diversification of Malvaceae in the Northern Hemisphere. The central Tibetan Plateau was at a low elevation with a monsoon influence during the Eocene, allowing the development of a subtropical flora containing Malvaceae. The taxonomic study of fossils from the Eocene of what is now the Tibetan Plateau is still ongoing. Malvaceae fossils from the Eocene Jianglang flora, are attributed to sub-families Tilioideae and Sterculioideae, and are compared with modern species. A new specimen of Firmiana is described based on a fruit valve with a pinnate venation, the secondary veins starting at the ventral suture and reaching the midvein, and the seeds attached at the proximal part of the ventral suture. This specimen represents the earliest known occurrence of the genus. A new occurrence of Craigia is also reported based on detached membranous valves of a fruit capsule with a prominent fusiform locular area and radiating venation. Based on the fossil record of Firmiana and its modern distribution, we infer that the genus may have originated in East Asia and subsequently diversified in South China and Southeast Asia. The new occurrence of Craigia indicates that the genus was distributed in humid areas in South, Southwest and North China during the Eocene. Both fossil records evidence the important role that the Tibetan region played in the diversification of plants in East and Southeast Asia.












