



Professor Wen-Tsai Wang (王文采, June 5, 1926–November 16, 2022) was an academician of the Chinese Academy of Sciences (CAS) and a legendary plant taxonomist at the Institute of Botany of CAS (Fig. 1). Herein, we organize a virtual special issue in Journal of Systematics and Evolution (JSE) to celebrate the legacy and life of Professor Wang, who was a leading plant taxonomist in China and made important contributions toward advancing the understanding of the flora of China, the biogeography of eastern Asia, and biodiversity research in the vast Hengduan Mountains. He served as the Editor-in-Chief of Acta Phytotaxonomica Sinica (now JSE) for 6 years from 1982 to 1988, and trained several generations of plant taxonomists in China (Li, 2001).
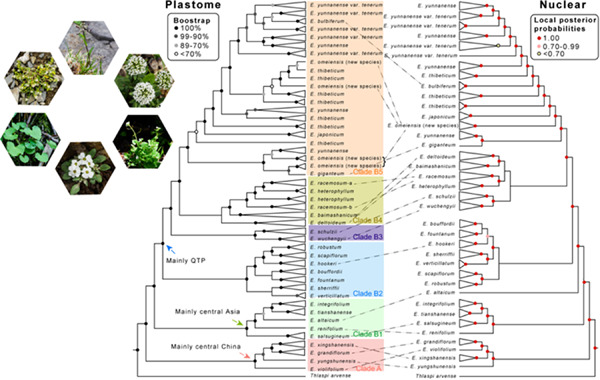
Both geographic isolation and polyploidization are assumed to play an important role in driving species diversification. However, this is rarely illustrated through phylogenomic analyses. The genus Eutrema (Brassicaceae), which also includes the salt-resistant species, are distributed mainly in Asia with extensive species diversification in the Qinghai–Tibet Plateau (QTP) and adjacent regions. In this study, we revealed almost fully resolved backbone relationships of the genus with genome re-sequencing data for genomes of 168 individuals from 28 species. Phylogenetic analyses of both plastomes and single-copy nuclear genes from the whole genome recovered six well-supported clades with almost consistent relationships. The first two clades are mainly distributed in central China and central Asia, while the other four in the QTP and adjacent regions. All of them diversified within 12 million years. Within each clade, we recovered numerous conflicts in the interspecific relationships between nuclear and plastome phylogenies, likely suggesting hybridization and incomplete lineage sorting during species diversification. Our estimation of genome size and comparison of the number of the single-copy nuclear genes demonstrated frequent occurrences of polyploids in the genus. Except for an establishment of the backbone phylogeny, our phylogenomic analyses suggest that in addition to strong geographic isolation, polyploidization may have played an important role in species diversification of this genus.
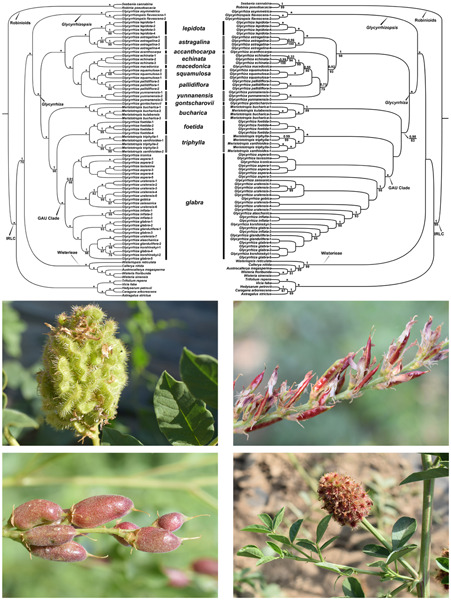
The liquorice tribe Glycyrrhizeae is a leguminous herbaceous group of plants comprised of the genera Glycyrrhiza and Glycyrrhizopsis. Some Glycyrrhiza taxa contain glycyrrhizin, a pharmacologically significant sweet substance that also has applications in crafting industrial materials. Here, we utilized an expanded taxon sampling of Glycyrrhizeae to reconstruct the phylogenetic relationships in the tribe based on genome skimming data, including whole chloroplast genomes, nuclear ribosomal DNA, and low-copy nuclear DNA. We also launched machine learning analysis (MLA) for one species pair with controversial taxonomic boundary. The integrated results indicated Glycyrrhizopsis should be split from Glycyrrhiza, while the former genus Meristotropis should be treated as part of Glycyrrhiza. Glycyrrhizopsis includes two species, Glycyrrhizopsis asymmetrica and Glycyrrhizopsis flavescens, and we recognize 13 species in Glycyrrhiza: Glycyrrhiza acanthocarpa, Glycyrrhiza astragalina, Glycyrrhiza bucharica, Glycyrrhiza echinata, Glycyrrhiza foetida, Glycyrrhiza glabra, Glycyrrhiza gontscharovii, Glycyrrhiza lepidota, Glycyrrhiza macedonica, Glycyrrhiza pallidiflora, Glycyrrhiza squamulosa, Glycyrrhiza triphylla, and Glycyrrhiza yunnanensis. We propose a broader G. glabra that includes former Glycyrrhiza aspera, G. glabra s.s., Glycyrrhiza inflata, and Glycyrrhiza uralensis, and represents the glycyrrhizin-contained medicinal group. Our ancestral state inferences show the ancestor of Glycyrrhiza lacked glycyrrhizin, and the presence of glycyrrhizin evolved twice within Glycyrrhiza during the last one million years. Our integrative phylogenomics-MLA study not only provides new insights into long-standing taxonomic controversies of Glycyrrhizeae, but also represents a useful approach for future taxonomic studies on other plant taxa.

The taxonomy and systematics of Urgineoideae (Hyacinthaceae) have been controversial in recent decades, with contrasting taxonomic treatments proposed based on preliminary and partial studies that have focused on morphology and/or solely plastid DNA sequence data. Some authors have recognized only two genera, with a very broadly conceived Drimia, while others have accepted several genera that, although better defined morphologically, were doubtfully monophyletic. Here, we present phylogenetic analyses involving four plastid DNA regions (trnL intron, trnL-F spacer, matK, and the trnCGCA-ycf6 intergenic region), a nuclear region (Agt1), and a selection of 40 morphological characters. Our study covers 293 samples and ca. 160 species of Urgineoideae (ca. 80% of its global diversity). Bayesian inference, maximum likelihood, and maximum parsimony analyses were performed to derive the phylogenetic patterns. The combination of data yielded phylogenetic trees with 31 well-defined clades or lineages, most corresponding to previously described genera, although some have required description or revised circumscription. As with other monocot families, a considerable degree of homoplasy was observed in morphological characters, especially in those groups with unspecialized flowers; nonetheless, consistent syndromes of traditional and novel characters are shown to support clade recognition at genus rank. The forthcoming revised classification of Urgineoideae is outlined here.
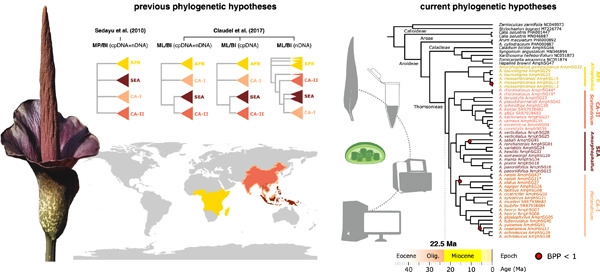
Encompassing ca. 200 species distributed in paleotropical Africa and Asia, Amorphophallus is one of the largest genera of Araceae. In spite of the great economic interest in its glucomannan production, only a few studies have attempted to grasp the evolutionary history of this genus. In the current state of knowledge, four main clades, mostly linked to biogeographical delineation, have been identified from phylogenies based on a few genes. However, relationships among and within these clades still remain unclear, due to the rapid radiation that occurred during the early evolutionary history of the genus. Here, we generated genome skimming libraries for 43 specimens from 36 species distributed across the 4 clades, which allowed us to produce a phylogenetic matrix for a set of 71 plastid genes. Our phylogenies confirm the monophyly of these clades but show a new and well-resolved arrangement among these clades. Our analyses therefore provide a new scenario and timeline for the evolution of the main Amorphophallus clades, consistent with the morphological characteristics of the clades. The inferred scenario is also in agreement with climate dynamics and the onset of long-distance dispersal by the earliest migratory birds near the Oligocene/Miocene transition around 23 million years ago. Our study provides an up-to-date baseline to understand biogeographic and ecological processes that shaped the current diversity and distribution of Amorphophallus, paving the way for larger-scale phylogenomic studies based on plastid and nuclear genomes.
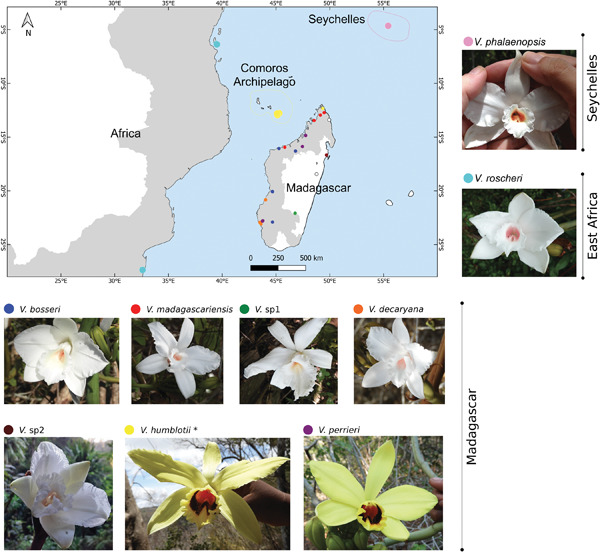
The leafless Vanilla species complex from the South-West Indian Ocean (SWIO) region has long been a taxonomic challenge, due to limited patterns of morphological differentiation and an absence of variation within chloroplast sequences. This complex includes seven known morphospecies: V. madagascariensis, V. bosseri, V. decaryana, and V. perrieri endemic to Madagascar, V. humblotii presumed as endemic to the Comoros Archipelago, but also present in Madagascar, V. roscheri from the East African coast, and V. phalaenopsis endemic to Seychelles. A previous population genetic study using microsatellite markers allowed us to distinguish, in addition to the five recognized Malagasy taxa, two other genetic clusters present in the East of the island. An integrative taxonomy approach was therefore conducted by combining microsatellite and morphological data used in the previous study with new data sets, and by adding ITS sequencing data, to validate the taxonomic level of these Malagasy genetic clusters and unravel phylogenetic relationships between SWIO species. As a result, based on phylogenetic, genotypic and morphological evidence, nine species were discriminated in the SWIO region, including seven in Madagascar, with two new eastern species. The leafless Vanilla group originated and diversified in Madagascar, from an ancestor of African descent, with three subsequent independent colonization events from Madagascar to the other territories of SWIO within the two main lineages (white versus yellow flower species). The new Malagasy species, V. allorgeae Andriamihaja & Pailler sp. nov., and V. atsinananensis Andriamihaja & Pailler sp. nov., are described and a new identification key is proposed.

Oresitrophe and Mukdenia (Saxifragaceae) are epilithic sister genera used in traditional Chinese medicine. The taxonomy of Mukdenia, especially of M. acanthifolia, has been controversial. To address this, we produced plastid and mitochondrial data using genome skimming for Mukdenia acanthifolia and Mukdenia rossii, including three individuals of each species. We assembled complete plastomes, mitochondrial CDS and nuclear ribosomal ETS/ITS sequences using these data. Comparative analysis shows that the plastomes of Mukdenia and Oresitrophe are relatively conservative in terms of genome size, structure, gene content, RNA editing sites and codon usage. Five plastid regions that represent hotspots of change (trnH-psbA, psbC-trnS, trnM-atpE, petA-psbJ and ccsA-ndhD) are identified within Mukdenia, and six regions (trnH-psbA, petN-psbM, trnM-atpE, rps16-trnQ, ycf1 and ndhF) contain a higher number of species-specific parsimony-informative sites that may serve as potential DNA barcodes for species identification. To infer phylogenetic relationships between Mukdenia and Oresitrophe, we combined our data with published data based on three different datasets. The monophyly of each species (Oresitrophe rupifraga, M. acanthifolia and M. rossii) and the inferred topology ((M. rossii, M. acanthifolia), O. rupifraga) are well supported in trees reconstructed using the complete plastome sequences, but M. acanthifolia and M. rossii did not form a separate clade in the trees based on ETS + ITS data, while the mitochondrial CDS trees are not well-resolved. We found low recovery of genes in the Angiosperms353 target enrichment panel from our unenriched genome skimming data. Hybridization or incomplete lineage sorting may be the cause of discordance between trees reconstructed from organellar and nuclear data. Considering its morphological distinctiveness and our molecular phylogenetic results, we strongly recommend that M. acanthifolia be treated as a distinct species.
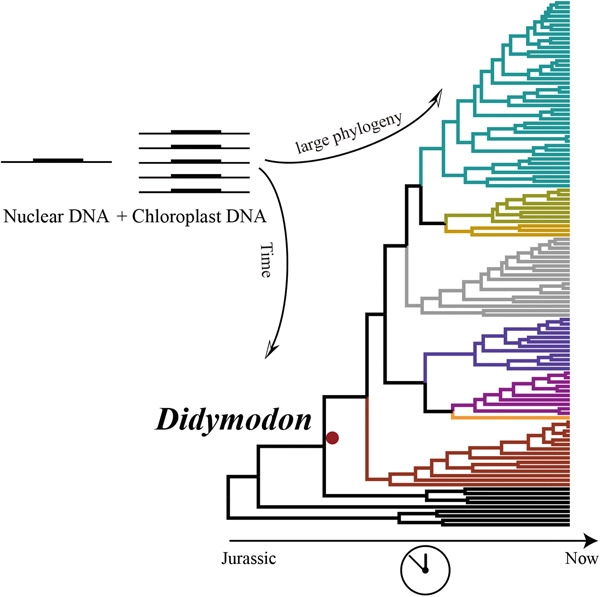
Didymodon Hedw., with approximately 140 species in the family Pottiaceae, is distributed nearly throughout the world, with the greatest diversity and important ecological functions in drought lands and alpine ecosystems. Several studies involving morphology, molecular systematics, and macro-systematic analysis have addressed the infrageneric classification of Didymodon, but controversy over the position of the infrageneric and species classification remains due to its high degree of morphological variation in micro-habitats and strong sensitivity to climate change at regional and global scale. To date, only a few phylogenetic studies have been conducted with an incomplete number of Didymodon species; further, there is no study published regarding the divergence time of Didymodon. Consequently, we conducted a comprehensive phylogenetic analysis of Didymodon species, sampling a total of 107 species, based on one nuclear (ITS) and five chloroplast DNA. Moreover, divergence time analysis was conducted to infer the age of origin and divergence of Didymodon species. Our results presented the largest scale phylogenetic relationship of Didymodon to date and resolved the phylogenetic status of some controversial taxa and the new species. The divergence time estimation showed that Didymodon species originated around the early Cretaceous, and the diversification was concentrated in the Cretaceous and Eocene. Paleoclimate and environmental change have a direct impact on the origin and divergence of Didymodon species by shaping their morphology, resource availability and ecological niche. Our study will help understand species origin and speciation of Didymodon as well as reflecting species adaptability and experience to historical events.
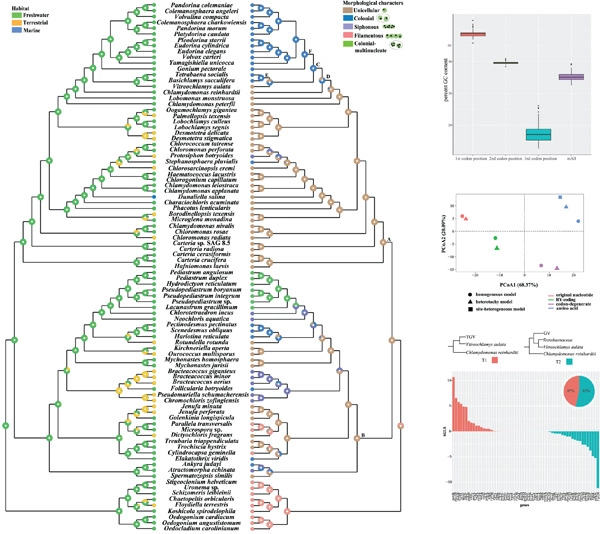
Volvocales forms a species-rich clade with wide morphological variety and is regarded as an ideal model for tracing the evolutionary transitions in multicellularity. The phylogenetic relationships among the colonial volvocine algae and its relatives are important for investigating the origin of multicellularity in the clade Reinhardtinia. Therefore, a robust phylogenetic framework of the unicellular and colonial volvocine algae with broad taxon and gene sampling is essential for illuminating the evolution of multicellularity. Recent chloroplast phylogenomic studies have uncovered five major orders in the Chlorophyceae, but the family-level relationships within Sphaeropleales and Volvocales remain elusive due to the uncertain positions of some incertae sedis taxa. In this study, we contributed six newly sequenced chloroplast genomes in the Volvocales and analyzed a dataset with 91 chlorophycean taxa and 58 protein-coding genes. Conflicting phylogenetic signals were detected among chloroplast genes that resulted in discordant tree topologies among different analyses. We compared the phylogenetic trees inferred from original nucleotide, RY-coding, codon-degenerate, and amino acid datasets, and improved the robustness of phylogenetic inference in the Chlorophyceae by reducing base compositional bias. Our analyses indicate that the unicellular Chlamydomonas and Vitreochlamys are close to or nested within the colonial taxa, and all the incertae sedis taxa are nested within the monophyletic Sphaeropleales s.l. We propose that the colonial taxa in the Reinhardtinia are paraphyletic and multicellularity evolved once in the volvocine green algae and might be lost in Chlamydomonas and Vitreochlamys.

Endemic species are important components of regional biodiversity and hold the key to understanding local adaptation and evolutionary processes that shape species distributions. This study investigated the biogeographic history of a relict conifer Pinus bungeana Zucc. ex Endl. confined to central China. We examined genetic diversity in P. bungeana using genotyping-by-sequencing and chloroplast and mitochondrial DNA markers. We performed spatial and temporal inference of recent genetic and demographic changes, and dissected the impacts of geography and environmental gradients on population differentiation. We then projected P. bungeana's risk of decline under future climates. We found extremely low nucleotide diversity (average π 0.0014), and strong population structure (global FST 0.234) even at regional scales, reflecting long-term isolation in small populations. The species experienced severe bottlenecks in the early Pliocene and continued to decline in the Pleistocene in the western distribution, whereas the east expanded recently. Local adaptation played a small (8%) but significant role in population diversity. Low genetic diversity in fragmented populations makes the species highly vulnerable to climate change, particularly in marginal and relict populations. We suggest that conservation efforts should focus on enhancing gene pool and population growth through assisted migration within each genetic cluster to reduce the risk of further genetic drift and extinction.
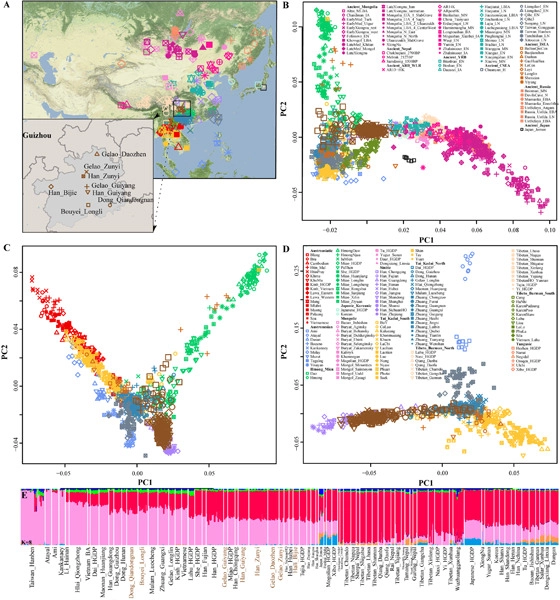
South China (SC) was a region with mixed rice–millet farming during the Middle Neolithic period and was also suggested to be the homeland of Tai-Kadai (TK)-speaking people. However, the formations of inland TK-speaking people and southwestern Hans are far from clear due to very few studies on this subject. Here, we reveal the spatiotemporally demographic history of SC by analyzing newly-generated genome-wide SNP data of 115 modern southwestern individuals and find that inland TK-speaking Dongs and Bouyeis have a close genomic affinity to coastal TK/Austronesian (AN)-speaking people and Neolithic Yangtze River basin (YZRB) farmers, while southwestern Hans and TK-speaking Gelaos possess a close genomic affinity to Neolithic Yellow River basin (YRB) farmers. Genetic differentiations are identified among TK people from SC and Southeast Asia, and between northern and southern inland Chinese TK people, in which the identified shared genetic ancestry between TK and AN people highlights a common origin of AN/TK groups. Conclusively, our findings indicate that millet farmers deriving from the YRB and rice farmers deriving from the YZRB substantially contribute to the present-day inland TK speakers and southwestern Hans via a two-way admixture scenario of bi-directional gene-flow events, which facilitates the formation of a modern two-way genetic admixture profile.
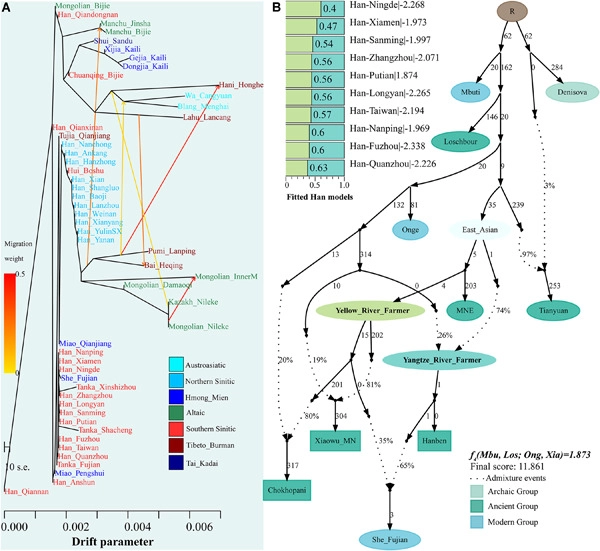
The population history of Southeast (SE) China remains poorly understood due to the sparse sampling of present-day populations and limited modeling with ancient genomic data. We report genome-wide genotyping data from 207 present-day Han Chinese and Hmong-Mien (HM)-speaking She people from Fujian and Taiwan Island, SE China. We coanalyzed 66 Early Neolithic to Iron Age ancient Fujian and Taiwan Island individuals obtained from previously published works to explore the genetic continuity and admixture based on patterns of genetic variations of the high-resolution time transect. We found the genetic differentiation between northern and southern East Asians was defined by a north–south East Asian genetic cline and our studied southern East Asians were clustered in the southern end of this cline. The southeastern coastal modern East Asians are genetically similar to other southern indigenous groups as well as geographically close to Neolithic-to-Iron Age populations, but they also shared excess alleles with post-Neolithic Yellow River ancients, which suggested a southward gene flow on the modern southern coastal gene pool. In addition, we identified one new HM genetic cline in East Asia with the coastal Fujian HM-speaking She localizing at the intersection between HM and Han clines. She people show stronger genetic affinity with southern East Asian indigenous populations, with the main ancestry deriving from groups related to southeastern ancient indigenous rice farmers. The southeastern Han Chinese could be modeled with the primary ancestry deriving from the group related to the Yellow River Basin millet farmers and the remaining from groups related to rice farmers, which was consistent with the northern China origin of modern southeastern Han Chinese and in line with the historically and archaeologically attested southward migrations of Han people and their ancestors. Our estimated north–south admixture time ranges based on the decay of the linkage disequilibrium spanned from the Bronze Age to historic periods, suggesting the recent large-scale population migrations and subsequent admixture participated in the formation of modern Han in SE Asia.
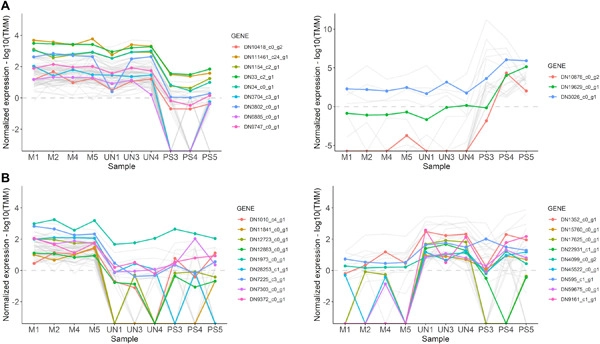
The environment is a powerful selective pressure for sessile organisms, such as plants, and adaptation to the environment is particularly important for long-lived species, like trees. Despite the importance of adaptive trait variation to the survival and success of trees, the molecular basis of adaptation is still poorly understood. Gene expression patterns in three closely related, but phenotypically and ecologically divergent, pine species were analyzed to detect differentiation that may be associated with their adaptation to distinct environments. Total RNA of Pinus mugo, Pinus uncinata, and Pinus sylvestris samples grown under common garden conditions was used for de novo transcriptome assembly, providing a new reference dataset that includes species from the taxonomically challenging P. mugo complex. Gene expression profiles were found to be very similar with only 121 genes significantly diverged in any of the pairwise species comparisons. Functional annotation of these genes revealed major categories of distinctly expressed transcripts, including wood trait properties, oxidative stress response, and response to abiotic factors such as salinity, drought, and temperature. We discuss putative associations between gene expression profiles and adaptation to different environments, for example, the upregulation of genes involved in lignin biosynthesis in the species, which have adapted to mountainous regions characterized by strong winds and thick snow cover. Our study provides valid candidates for verification of the importance of the gene expression role, in addition to evidence for selection within genomic regions, in the process of ecological divergence and adaptation to higher altitudes in pine taxa.
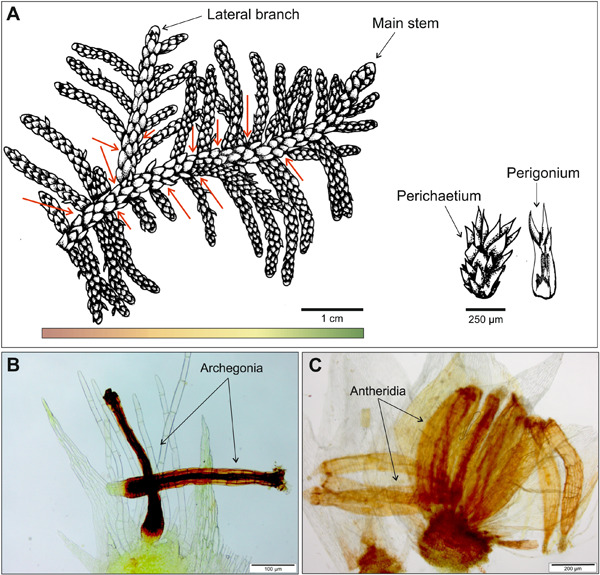
The impact of climate change on biodiversity operates through a complex mixture of habitat loss and range shift through the emergence of newly suitable areas (Warren et al., 2013). The main question is therefore to determine whether species have the ability to balance the loss of suitable habitats by effectively shifting their ranges and track suitable areas under climate change (Nogués-Bravo et al., 2018). Zanatta et al. (2020) most recently simulated the dispersal of apparently extremely efficient dispersers, namely bryophytes, whose tiny spores (<20 µm on average) are wind-dispersed across large distances, under several climate change scenarios. They concluded that, despite their high dispersal capacities, bryophytes will lose suitable areas at a faster rate than they will colonize newly suitable areas. Paradoxically, mounting evidence points to striking range expansions in epiphytic floras in the context of the sharp decrease of SO2 concentrations since the 1980s and climate warming (Tuba et al., 2011). Here, we addressed this apparent controversy by reassessing the results of Zanatta et al. (2020) in the light of the repartitioning of the data per habitat type.
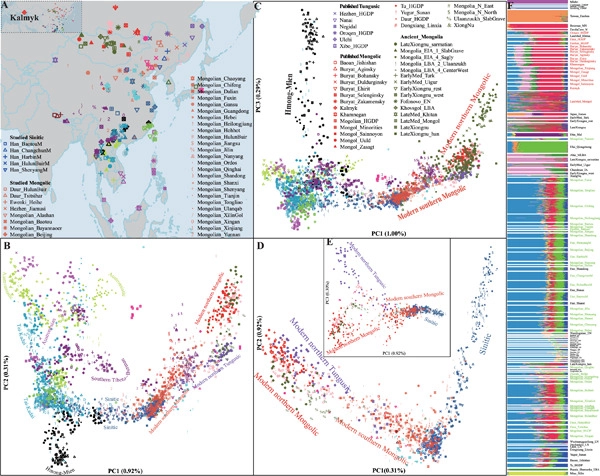
North China and South Siberia, populated by Altaic- and Sino-Tibetan-speaking populations, possess extensive ethnolinguistic diversity and serve as the crossroads for the initial peopling of America and western–eastern transcontinental communication. However, the population genetic structure and admixture history of northern East Asians remain poorly understood due to a lack of genome-wide data, especially for Mongolic-speaking people in China. We genotyped genome-wide single nucleotide polymorphisms for 510 individuals from 38 Mongolic, Tungusic, and Sinitic-speaking populations. We first explored the shared alleles and haplotypes within the studied groups. We then merged with 3508 published modern and ancient Eurasian individuals to reconstruct the deep evolutionary and natural selection history of northern East Asians. We identified genetic substructures within Altaic-speaking populations: Western Turkic people harbored more western Eurasian-related ancestry; Northern Mongolic people in Siberia and eastern Tungusic people in Amur River Basin (ARB) possessed a majority of Neolithic ARB related ancestry; Southern Mongolic people in China possessed apparent genetic influence from Neolithic Yellow River Basin (YRB) farmers. Additionally, we found the differentiated admixture history between western and eastern Mongolians and geographically close Northeast Hans: the former received a genetic impact from western Eurasians, and the latter retained the primary Neolithic YRB and ARB ancestry. Moreover, we demonstrated that Kalmyk people from the northern Caucasus Mountains possessed a strong genetic affinity with Neolithic Mongolian Plateau (MP) people, supporting the hypothesis of their eastern Eurasian origin and long-distance migration history. We also illuminated that historical pastoral empires in the MP contributed considerably to the gene pool of northern Mongolic people but rarely to the southern ones. We finally found natural selection signatures in Mongolians associated with alcohol metabolism. Our results demonstrated that the Neolithic ancestral sources from the MP or ARB played an important role in spreading Altaic populations and languages. The observed multisources of genetic diversity contributed significantly to the extensive ethnolinguistic diversity in northern East Asia.
The Journal of Systematics and Evolution would like to acknowledge and thank the following reviewers for their contributions in the period January 1–December 31 in 2022:
Akita, Shingo
Allen, Geraldine
An, Xin-Min
Appelhans, Marc
Bacci, Lucas
Bhardwaj, Pankaj
Brothers, Denis
Cai, Wan-Zhi
Cameron, Kenneth
Cardoso, Pedro
Cellinese, Nico
Chao, Zhi
Chen, Jin-Ming
Chen, Shi-Chao
Chen, Xiao-Yong
Chen, Zhi-Duan
Cheng, Feng
Chesters, Douglas
Cilli, Elisabetta
Corlett, Richard
Del Rio, Cédric
Denk, Thomas
Dering, Monika
Dillenberger, Markus
Domina, Gianniantonio
Draper, David
Du, Fang
Duckett, Jeffrey
Ebersbach, Jana
Edera, Alejandro
Edwards, Christine
Escudero, Marcial
Fan, Xing
Franco, Fernando
Fujiwara, Tao
Gao, Lian-Ming
Garnatje, Teresa
Ge, Song
Gibernau, Marc
Gillespie, Lynn
Gong, Yan-Bing
Guo, Xian-Guang
Guo, Ya-Long
He, Bin
Heřmanová, Zuzana
Herrera, Fabiany
Hetterscheid, Wilbert
Holstein, Norbert
Howard, Cody
Huang, Bin-Quan
Huang, Shuang-Quan
Huang, Ji-Hong
Huang, Xiao-Lei
Huang, Zheng-Zhong
Ickert-Bond, Stefanie
Ikeda, Hajime
Jia, Hui
Jiang, Ting-Lei
Jiang, Xiao-Long
Jiao, Heng-Wu
Jiao, Yuan-Nian
Jin, Xiao-Hua
Jin, Jian-Hua
Johnson, Steven D.
Jud, Nathan
Kandasamy, Dineshkumar
Kikuchi, Satoshi
Kim, Sang-Tae
Kociolek, John
Koenemann, Daniel
Köhler, Matias
Kong, Xiang-Bo
Kunzmann, Lutz
Laffan, Shawn
Leslie, Andrew
Li, Hong-Tao
Li, Jianhua
Li, Jia-Tang
Li, Lang
Li, Lin-Feng
Li, Lu-Lu
Li, Ming-He
Li, Pan
Li, Xin-Xin
Liao, Wan-Jin
Lin, Ai-Qing
Liu, Dong
Liu, Hong-Mei
Liu, Shih-Hui
Liu, Tong-Yi
Liu, Zhi-Yong
Loizeau, Pierre-André
Long, Chun-Lin
Lu, Li-Min
Luo, A-Rong
Luo, Yi-Bo
Lyskov, Dmitry
Ma, Tao
Mandel, Jennifer
Manen, Jean-François
Mao, Jian-Feng
Mao, Kang-Shan
Mao, Ling-Feng
Marques, Isabel
McAdam, Scott
Mi, Xiang-Cheng
Michelangeli, Fabian
Mishler, Brent
Mishra, Geetanjali
Morgan, John
Múlgura, Maria Ema
Nickrent, Daniel
Nicolas, Antoine
Nie, Ze-Long
Olofsson, Jill
Olsson, Sanna
Park, Seonjoo
Patrizia, Serventi
Piwczyński, Marcin
Qiu, Ying-Xiong
Raguso, Robert
Ramos, Sergio
Ran, Jin-Hua
Rebollo, Roberto
Ren, Zong-Xin
Rojas-Andrés, Blanca
Rose, Jeffrey
Saavedra, Serguei
Salazar, Gerardo
Sanchez Reyes, Luna Luisa
Sawicki, Jakub
Schlueter, Philipp
Schneider, Harald
Shi, Gong-Le
Shi, Su-Hua
Shi, Wei
Stökl, Johannes
Stuessy, Tod
Su, Tao
Suda, Shoichiro
Sun, Hong-Zheng
Sun, Miao
Sundue, Michael
Tan, Yun-Hong
Tang, Liang
Tang, Long
Tang, Qing
Tembrock, Luke
Tomasello, Salvatore
Tribble, Carrie
Tu, Tie-Yao
Valcarcel, Virginia
Varga, Sandra
Vatanparast, Mohammad
Wan, Justin S. H.
Wang, Ai-Ying
Wang, Bao-Sheng
Wang, Gang
Wang, Geoff
Wang, Heng-Chang
Wang, Hong-Wei
Wang, Jing
Wang, Mao-Jun
Wang, Nian
Wang, Wei
Wang, Yin-Zheng
Wang, Ze-Fu
Watanabe, Shin
Weng, Mao-Lun
Wu, Yun-Ke
Wu, Zeng-Yuan
Wu, Zhi-Qiang
Xiang, Chun-Lei
Xiang, Qiao-Ping
Xiang, Qiu-Yun (Jenny)
Xiang, Xiao-Guo
Xie, San-Ping
Xie, Shu-Lian
Xing, Yao-Wu
Xu, Jian-Hong
Xu, Xiao-Ting
Xue, Huai-Jun
Yan, Yu-Jing
Yan, Yue-Hong
Yang, Melinda
Yang, Shi-Xiong
Ye, Hang
Ye, Jian-Fei
Yi, Xueling
Yoon, Hwan Su
Yu, Xiang-Qin
Yu, Yan
Yu, Yi-Lun
Zander, Richard
Zhang, Zhi-Qiang
Zhang, Chi
Zhang, Hai-Qing
Zhang, Ling
Zhang, Qun-Jie
Zhang, Xian-Chun
Zhang, Xiao-Ming
Zhang, Xiao-Xia
Zhang, Xu
Zhao, Liang
Zhao, Yi-Yong
Zhou, Ren-Chao
Zhou, Zhi-Jun
Zhu, An-Dan
Zou, Zheng-Ting
Zu, Pengjuan












