Huijuan Zhou, Fan Wu, Hengzhao Liu, Jiayu Ma, Huiling Yan, Renna Li, Lu Fan, Fangbing Ding, Yuwei Linghu, Bin Xie, Xiaoai Fang, Shu Yang, Ming Yue, Peng Zhao, Yaling Wang
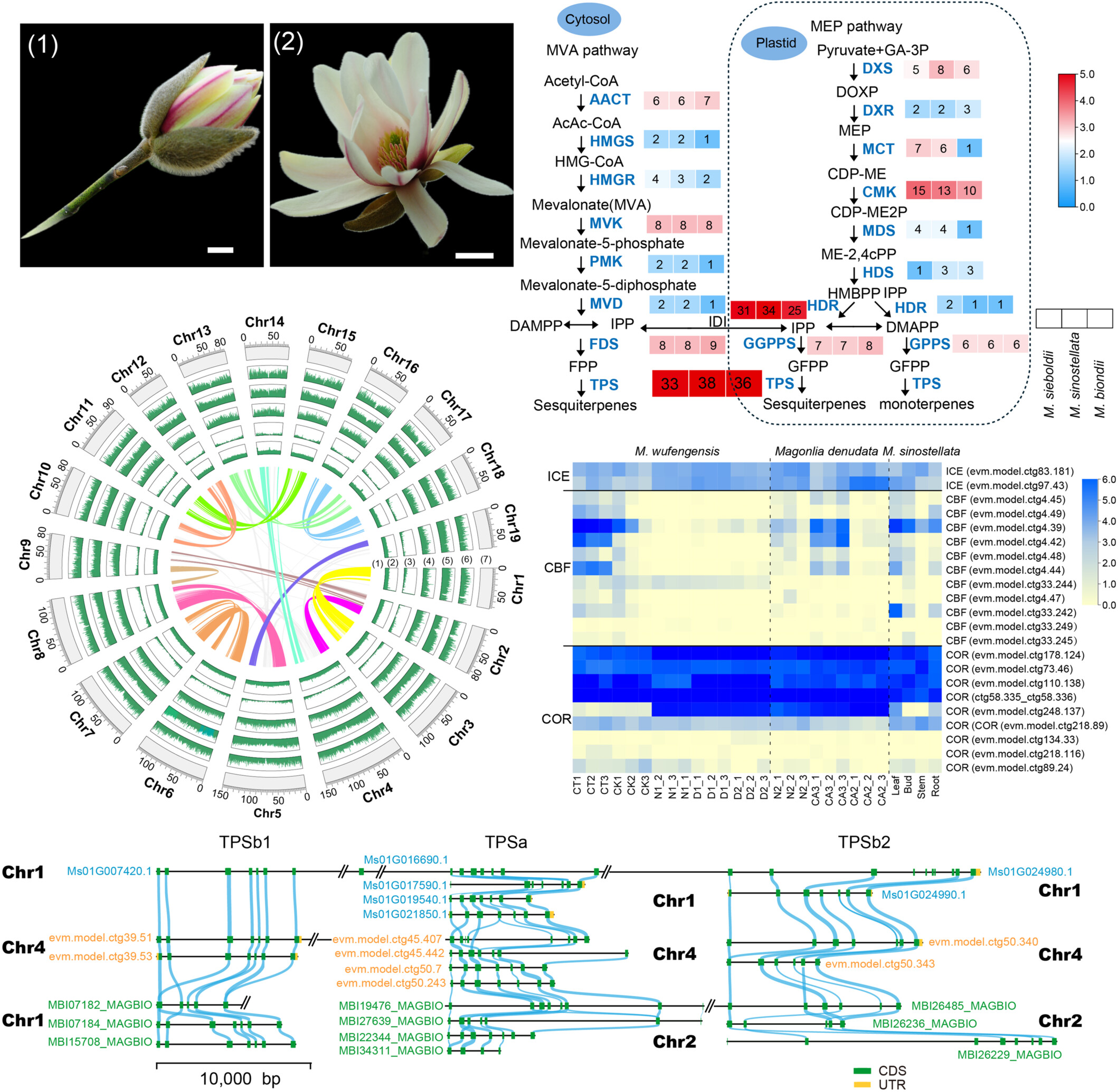
The genus Magnolia belongs to Magnoliaceae, an early diverging lineage of the Magnoliales, and is cultivated globally for its high ornamental and commercial values. As a large genus in the family Magnoliaceae, Magnolia species are regarded as highly valuable in phylogenetic and conservation biological studies. However, the whole genome data of Magnolia is still relatively insufficient. Here, we present a high-quality, chromosome-level genome sequence of Magnolia sinostellata (1.86 Gb) with a scaffold N50 of 85.33 Mb. The 19 M. sinostellata genome chromosomes revealed 11 main duplications representing the subgenome. Comparative genomics analysis revealed that the variance in the number of abiotic stress resistance genes among Magnoliid species are related to different environmental adaptations. Most of the genes related to MAPK signaling and stress resistance pathways in the investigated M. sinostellata species are expanded, compared to the other species. Furthermore, the comparative genomics analysis of three Magnolia assemblies, M. sinostellata, Magnolia biondii, and Magnolia sieboldii revealed that large inversions were enriched in terpenoid metabolic pathways, stress resistance and flavonoid biosynthesis, and DNA replication proteins. Using transcriptome sequencing data, we analyzed the expression levels of genes related to terpenoid biosynthesis (terpene synthase) and ICE–CBF–COR gene models related to cold tolerance in various tissues and the buds under different temperature conditions. The high-quality assembly of M. sinostellata and the ICE–CBF–COR bioinformatic analysis cascade provide valuable resources for studying the phylogeny and evolution of Magnoliaceae and angiosperms, while the candidate genes will provide foundational support for molecular breeding in Magnolia species.
A valuable genomic resource for understanding the evolution, stress resistance, and terpenoid biosynthesis in Magnolia sinostellata. The high-quality genome assembly and detailed analysis offer insights into the adaptive evolution of this endangered species and lay the foundation for future conservation and molecular breeding efforts. The findings highlight the importance of M. sinostellata as a model for studying the evolutionary dynamics and functional genomics of the Magnoliaceae family.