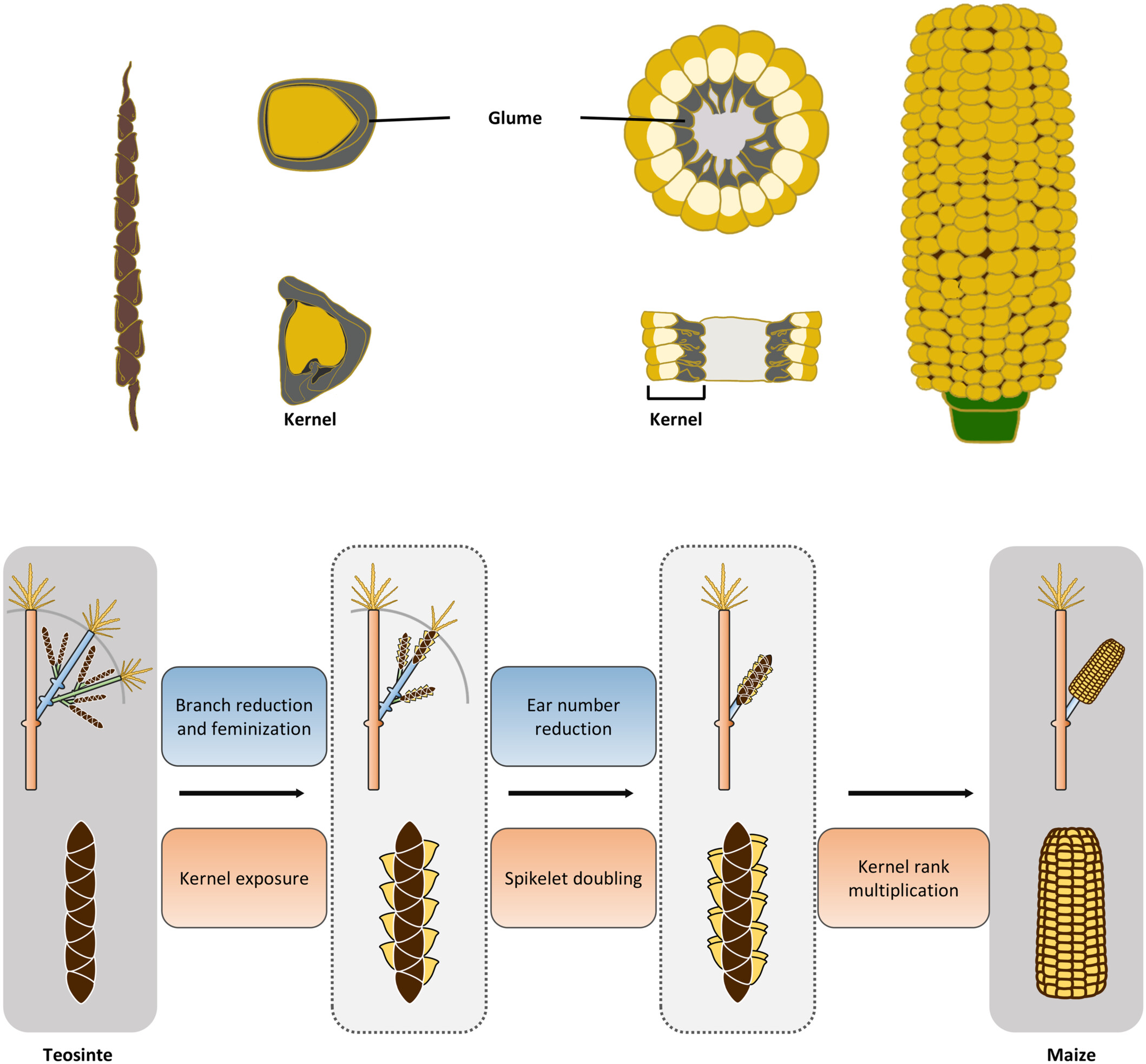
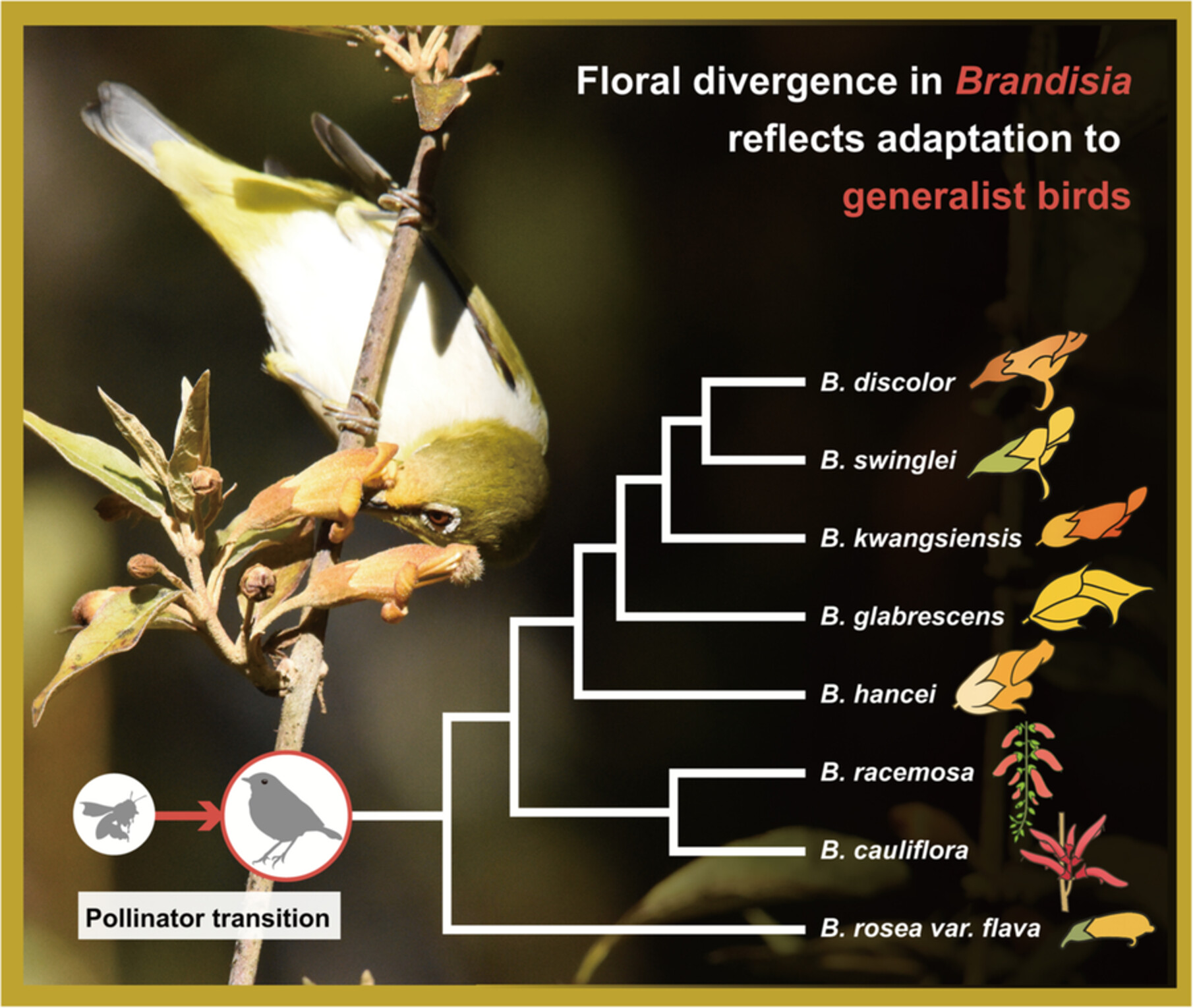
Zhe Chen, Chang-Qu Liu, Zi-Jue Ren, Hang Sun, Yang Niu
Pollinators are key drivers of floral evolution and diversification. While floral traits often converge in response to shared pollinators, they may diverge following pollinator shifts. Here, we examine the evolution of floral traits in Brandisia, a hemiparasitic genus endemic to East and Southeast Asia that shows notable interspecific variation. We combined field observations, a literature survey of pollination systems across Orobanchaceae, measurements of 14 floral traits, and phylogenetically informed comparative analyses. Our results show that Brandisia species are primarily bird-pollinated, probably derived from the bee-pollinated condition predominant in Orobanchaceae. Their flowers show typical bird-pollination traits, including tubular corollas, exserted reproductive organs, and abundant dilute nectar. Several traits may also function to avoid antagonists through visual (e.g., red coloration inconspicuous to bees), morphological (e.g., reduced or recurved corolla lobes), or physiological (e.g., dilute nectar) barriers. Ancestral state reconstruction indicates that the common ancestor of Brandisia had moderately specialized floral traits, including solitary axillary flowers, orange-yellow coloration, short tubular corollas, and hexose-dominated nectar. From this ancestral condition, both more specialized and more generalized phenotypes evolved, involving 11 shifts across eight traits. Together, our findings indicate that Brandisia is predominantly bird-pollinated in the Asian flora. Rather than resulting from major pollinator shifts, floral trait variation in Brandisia reflects a continuum of adaptation to bird pollinators, potentially shaped by fine-scale niche partitioning. Some floral traits may also have evolved under additional selective pressures, such as avoiding bees. This study advances understanding of how bird pollinators shape floral diversification in angiosperm.