

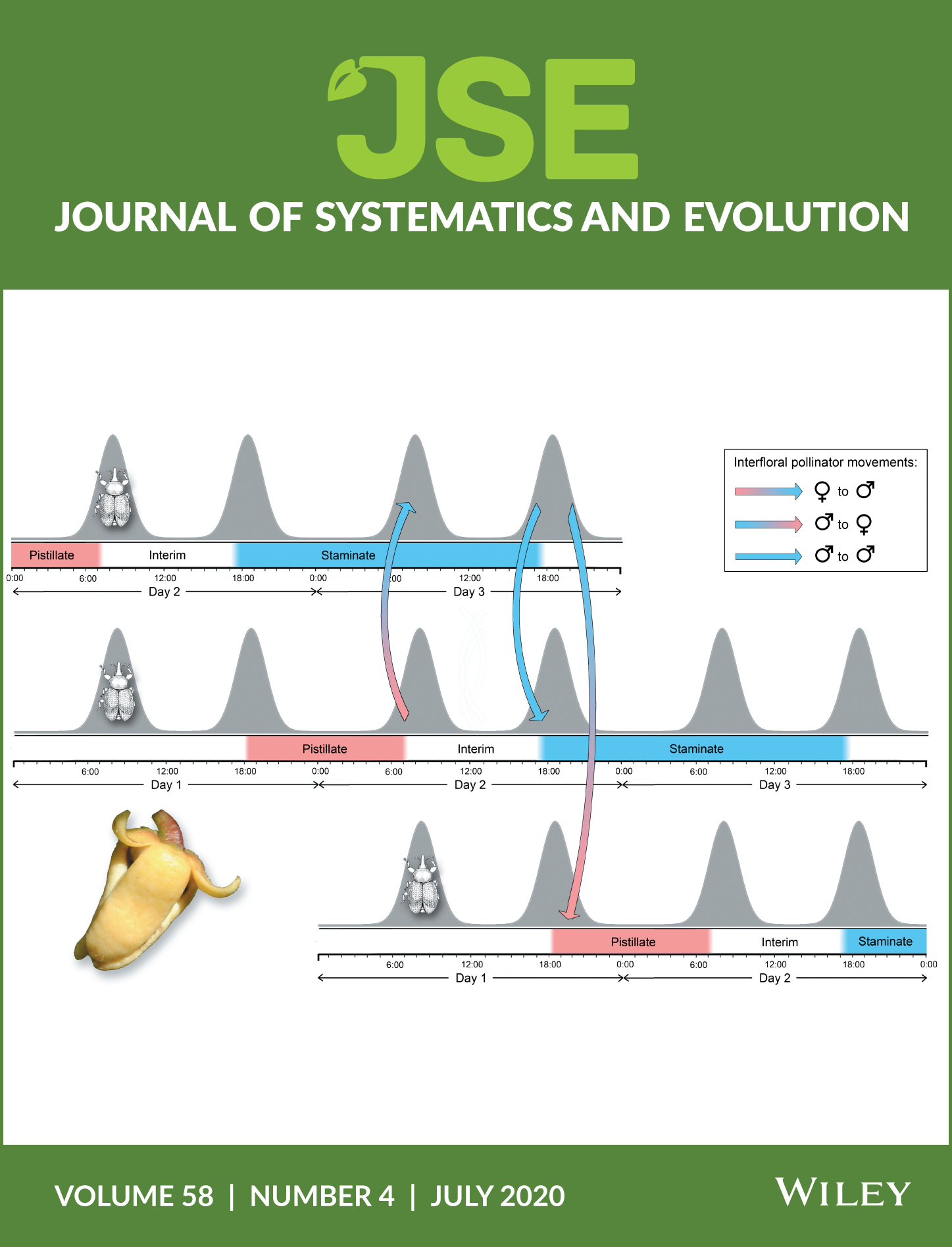
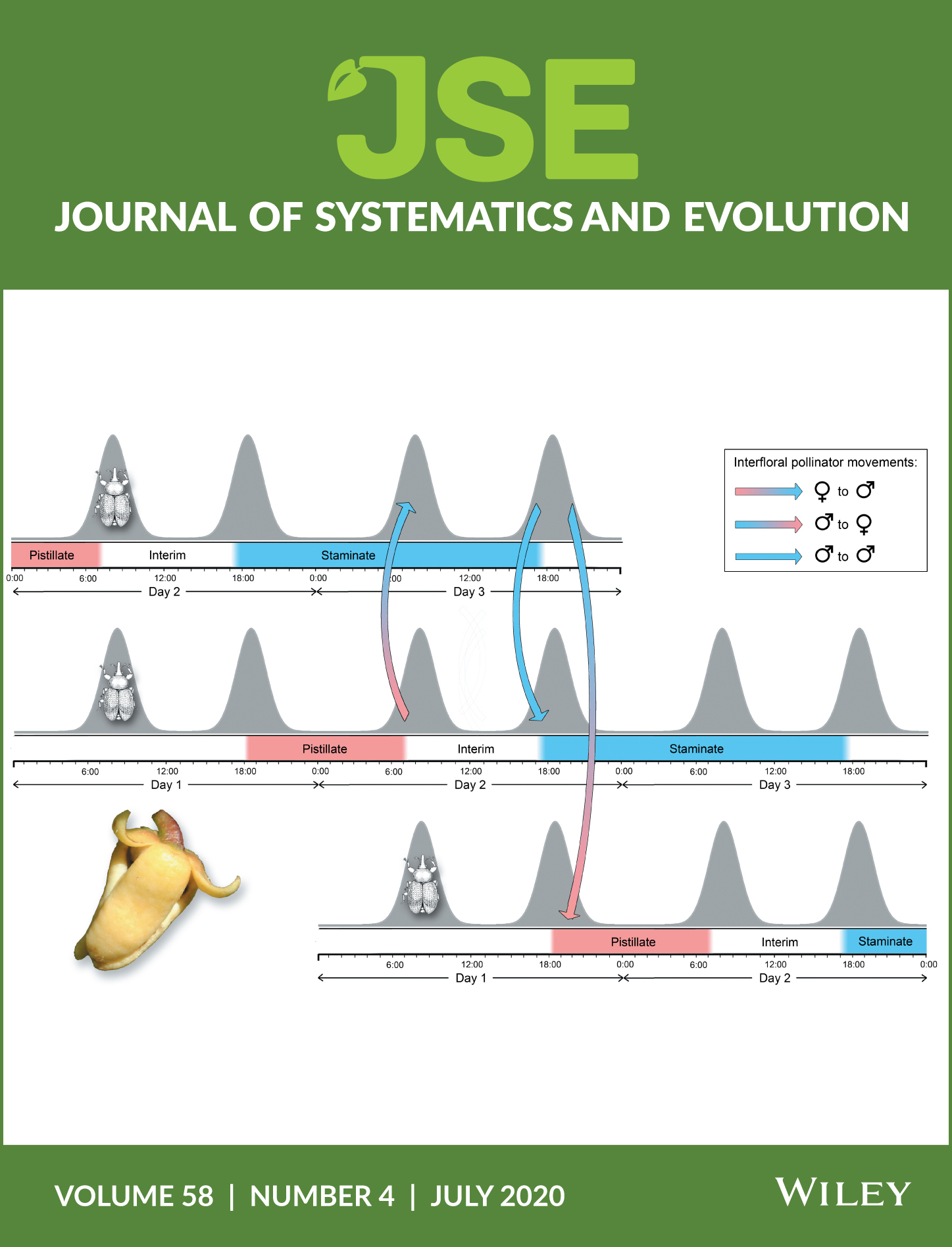
Potential key functional floral traits are assessed in the species‐rich early divergent angiosperm family Annonaceae. Pollinators (generally beetles) are attracted by various cues (particularly visual, olfactory, and thermogenic), with pollinators rewarded by nectar (generally as stigmatic exudate), heat, and protection within the partially enclosed floral chamber. Petals sometimes function as pollinator brood sites, although this could be deceptive. Annonaceae species are self‐compatible, with outcrossing promoted by a combination of protogyny, herkogamy, floral synchrony, and dicliny. Pollination efficiency is enhanced by pollen aggregation, changes in anthesis duration, and pollinator trapping involving a close alignment between petal movements and the circadian rhythms of pollinators. Most Annonaceae flowers are apocarpous, with syncarpy restricted to very few lineages; fertilization is therefore optimized by intercarpellary growth of pollen tubes, either by stigmatic exudate (suprastylar extragynoecial compitum) or possibly the floral receptacle (infrastylar extragynoecial compitum). Although Annonaceae lack a distinct style, the stigmas in several lineages are elongated to form “pseudostyles” that are hypothesized to function as sites for pollen competition. Flowers can be regarded as immature fruits in which the ovules are yet to be fertilized, with floral traits that may have little selective advantage during anthesis theoretically promoting fruit and seed dispersal. The plesiomorphic apocarpous trait may have been perpetuated in Annonaceae flowers as it promotes the independent dispersal of fruit monocarps (derived from separate carpels), thereby maximizing the spatial/temporal distance between seedlings. This might compensate for the lack of genetic diversity among seeds within fruits arising from the limited diversity of pollen donors.
North America is a large continent with extensive climatic, geological, soil, and biological diversity. As biota faces threat from habitat destruction and climate change, making a quantitative assessment of biodiversity becomes critically important. Rapid digitization of plant specimen records and accumulation of DNA sequence data enable a much‐needed broad synthesis of species occurrences with phylogenetic data. In this study, the first such synthesis of a flora from such a large and diverse part of the world is attempted, all seed plants from the North American continent (here defined to include Canada, United States, and Mexico), with a focus on examining phylogenetic diversity and endemism. We collected digitized plant specimen records and chose a coarse grain for analysis, recognizing that this grain is currently necessary for reasonable completeness per sampling unit. We found that raw richness and endemism patterns largely support previous hypotheses of biodiversity hotspots. The application of phylogenetic metrics and a randomization test revealed novel results, including a significant phylogenetic clustering across the continent, a striking east–west geographical difference in the distribution of branch lengths, and the discovery of centers of neo‐ and paleoendemism in Mexico, the southwestern USA, and the southeastern USA. Finally, our examination of phylogenetic beta diversity provides a new approach to compare centers of endemism. We discuss the empirical challenges of working at the continental scale and the need for more sampling across large parts of the continent, for both DNA data for terminal taxa and spatial data for poorly understood regions, to confirm and extend these results.
Taxonomy is a traditional subject, but it still receives attention and has become a topic of much discussion in recent years. Many of these discussions have raised concerns about the future of taxonomy, especially with regard to the workforce responsible for the discovery of new species in the context of declining biodiversity. Previous discussions were based on the taxonomic data of plants and animals, but the status of fungal taxonomy has not been mentioned. Fungi have one of the highest levels of biodiversity among all living organisms, second only to insects. The discussion of the future of taxonomy without the inclusion of fungal data is incomplete. Here, we present the results of analyses based on all new fungal taxa published since 1753. Fungal taxonomy is an ever‐growing area of study with increasing numbers of new taxa being described and growing numbers of fungal taxonomists. Compared with plants and most animal groups, there has been a much sharper increase in the rate at which new fungal taxa are being described. Furthermore, the number of taxonomists studying fungi has increased at a faster speed than those studying plants or animals. This indicates that fungal taxonomy is a prosperous subject and a dynamic area for scientific studies, and that it deserves much more attention and support. The study of fungal taxonomy will deepen our understanding of the biodiversity of our planet.
How to maximize the conservation of biodiversity is critical for conservation planning, particularly given rapid habitat loss and global climatic change. The importance of preserving phylogenetic diversity has gained recognition due to its ability to identify some influences of evolutionary history on contemporary patterns of species assemblages that traditional taxonomic richness measures cannot identify. In this study, we evaluate the relationship between taxonomic richness and phylogenetic diversity of angiosperms at genus and species levels and explore the spatial pattern of the residuals of this relationship. We then incorporate data on historical biogeography to understand the process that shaped contemporary floristic assemblages in a global biodiversity hotspot, Yunnan Province, located in southwestern China. We identified a strong correlation between phylogenetic diversity residuals and the biogeographic affinity of the lineages in the extant Yunnan angiosperm flora. Phylogenetic diversity is well correlated with taxonomic richness at both genus and species levels between floras in Yunnan, where two diversity centers of phylogenetic diversity were identified (the northwestern center and the southern center). The northwestern center, with lower phylogenetic diversity than expected based on taxonomic richness, is rich in temperate‐affinity lineages and signifies an area of rapid speciation. The southern center, with higher phylogenetic diversity than predicted by taxonomic richness, contains a higher proportion of lineages with tropical affinity and seems to have experienced high immigration rates. Our results highlight that maximizing phylogenetic diversity with historical interpretation can provide valuable insights into the floristic assemblage of a region and better‐informed decisions can be made to ensure different stages of a region's evolutionary history are preserved.
The family Lauraceae is a major component of tropical and subtropical forests worldwide, and includes some commercially important timber trees and medicinal plants. However, phylogenetic relationships within Lauraceae have long been problematic due to low sequence divergence in commonly used markers, even between morphologically distinct taxa within the family. Here we present phylogenetic analyses of 43 newly generated Lauraceae plastomes together with 77 plastomes obtained from GenBank, representing 24 genera of Lauraceae and 17 related families of angiosperms, plus nine barcodes from 19 additional species in 18 genera of Lauraceae, in order to reconstruct highly supported relationships for the Lauraceae. Our phylogeny supports the relationships: sisterhood of the Lauraceae and a clade containing Hernandiaceae and Monimiaceae, with Atherospermataceae and Gomortegaceae being the next sister groups, followed by Calycanthaceae. Our results highlight a monophyletic Lauraceae, with nine well‐supported clades as follows: Hypodaphnis clade, Beilschmiedia –Cryptocarya clade, Cassytha clade, Neocinnamomum clade, Caryodaphnopsis clade, Chlorocardium –Mezilaurus clade, Machilus –Persea clade, Cinnamomum –Ocotea clade, and Laurus –Neolitsea clade. The topology recovered here is consistent with the patterns of plastome structural evolution and morphological synapomorphies reported previously. More specifically, flower sex, living type, inflorescence type, ovary position, anther locus number, leaf arrangement, leaf venation, lateral vein number, tree height, and inflorescence location all represent morphological synapomorphies of different lineages. Our findings have taxonomic implications and two new tribes, Caryodaphnopsideae and Neocinnamomeae, are described, and the composition of four other tribes is updated. The phylogeny recovered here provides a robust phylogenetic framework through which to address the evolutionary history of the Magnoliids, the third‐largest group of Mesangiospermae.
Reconstructions of phylogenetic relationships in the flowering plant family Rubiaceae have up until now relied heavily on single‐ or multi‐gene data, primarily from the plastid compartment. With the availability of cost‐ and time‐efficient techniques for generating complete genome sequences, the opportunity arises to resolve some of the relationships that, up until now, have proven problematic. Here, we contribute new data from complete 58 plastid genome sequences, representing 55 of the currently 65 recognized tribes of the Rubiaceae. Also contributed are new data from the nuclear rDNA cistrons for corresponding taxa. Phylogenetic analyses are conducted on two plastid data sets, one including data from the protein coding genes only, and a second where protein coding data are combined with non‐coding regions, and on a nuclear rDNA data set. Our results clearly show that simply adopting a “more characters” approach does not resolve the relationships in the Rubiaceae. More importantly, we identify conflicting phylogenetic signals in the data. Analyses of the same plastid data, treated as nucleotides or as codon‐degenerated data, resolve and support conflicting topologies in the subfamily Cinchonoideae. As these analyses use the same data, we interpret the conflict to result from erroneous assumptions in the models used to reconstruct our phylogenies. Conflicting signals are also identified in the analyses of the plastid versus the nuclear rDNA data sets. These analyses use data from different genomic compartments, with different inheritance patterns, and we interpret the conflicts as representing “real” conflicts, reflecting biological processes of the past.
Dispersal scenarios have been favored over tectonic vicariance as an explanation for disjunct distributions in many plant taxa during the last two decades. However, this argument has been insufficiently addressed in cosmopolitan groups showing disjunct patterns in both the temperate and tropical regions. In this study, we used the Cannabaceae, an angiosperm family distributed in tropical and temperate regions of both the New World and the Old World, to explore the role of dispersal in shaping disjunct patterns and species diversification of cosmopolitan plants. We reconstructed the phylogenetic relationships of all 10 genera and 75 species of Cannabaceae (ca. 64.1% of recognized species) based on eight DNA regions. Based on fossil calibrations, we estimated the divergence times and net diversification rates. We further inferred the ancestral geographical ranges with several models and compared the fitness of different models. The Cannabaceae and most genera were strongly supported as monophyletic except for the Parasponia being embedded within the Trema . The Celtis were resolved into two strongly supported clades primarily corresponding to temperate and tropical regions. We inferred that the Cannabaceae originated at ca. 93 Ma, and that subsequent rampant and widespread dispersals shaped the intercontinentally disjunct distribution of the Cannabaceae. Dispersal coincides with adaptation to drier and colder climate in the Northern Hemisphere, or humid and warm climate in the tropical regions, followed by rapid species diversification. This study advances our understanding as to the formation of distribution patterns and species diversification of a plant family with tropical to temperate disjunct distributions.
The dwarf mountain pine (Pinus mugo ) and the Pyrenean pine (P. uncinata ) constitute a pair of closely related coniferous taxa of poorly resolved evolutionary history and affinity, which inhabit numerous stands scattered over subalpine environments of European mountain ranges. The aim of the study was to investigate their phylogeography and mutual relationships, shedding new light on their taxonomy and the past of the alpine flora. Previous evolutionary reconstructions of the mountain pines relied mainly on bi‐parentally or paternally inherited markers that quickly homogenize between populations, showing rather shallow and recent differentiation of gene pools. Therefore, to contrast these pictures, we analyzed diversity and differentiation within a large set of new mitochondrial loci, inherited in maternal line and distributed by seeds at short geographical distances. Samples of the taxa were taken from 27 natural populations representing their range‐wide distributions—17 populations of P. mugo and 10 of P. uncinata . All markers appeared polymorphic, providing a total of 31 multilocus haplotypes. Two of the loci proved to be species‐diagnostic and nearly fixed between analyzed samples. Distribution of mitotypes indicate that allopatric populations of the taxa constitute separate mitochondrial haplogroups, and the two mountain pines have independent evolutionary history. However, introgression of P. mugo mitotypes by P. uncinata specimens revealed in the species contact zone in Western Alps shows that their speciation is not fully completed.
Peripheral populations (i.e., those occurring on the edge of a species’ distribution range) can have different origins and genetic characteristics, and they may be critical for the conservation of genetic diversity. We investigated European peripheral populations of Scrophularia arguta , a widespread, annual plant distributed from Arabia to Northwest Africa and Macaronesia. Only two small disjunct population groups of this species occur in Europe, specifically in West‐Central and Southeast Iberia. To disclose the origin of these populations and determine their importance for the conservation of S. arguta genetic diversity, we analyzed DNA sequences from two nuclear and two plastid regions and amplified fragment length polymorphism markers in populations sampled mainly across the western distribution range of the species, and modeled the species distribution under present and late Quaternary conditions. The analyses revealed the presence of three distinct lineages of S. arguta in Europe, as a result of multiple colonization waves at different times in the Quaternary. Two of these lineages, occurring in Southeast Iberia, are the result of more or less recent dispersal from Northwest Africa. In contrast, West‐Central Iberian populations are strongly differentiated from the remaining range of S. arguta and can be considered as peripheral relict populations. Our study is the first to demonstrate the occurrence of at least three colonizations of the European continent from Africa by a native plant species. The diverse histories and genetic makeup of the resulting populations confirm the importance of peripheral populations, and particularly of ancient relict populations, for the conservation of global genetic diversity in widespread species.
Phytophthora cinnamomi (Pc ) is an extremely destructive soil‐borne pathogen of Asiatic origin responsible for “ink disease” in chestnut. This work assesses the adaptive potential to the impact of Pc of four Spanish populations of Castanea sativa undergoing different selection pressures. To explore the evolvability of C. sativa to Pc in the selected populations, parameters obtained from neutral and functional genetic diversity were compared with estimates of quantitative genetic variability. Nine expressed sequence tags‐simple sequence repeat (EST‐SSR) markers were selected and their transferability and polymorphism in 137 C. sativa individuals were evaluated. To test the potential of EST‐SSR markers for early selection of Pc tolerant plant material, the offspring of selected individuals were challenged with Pc . Expressed sequence tags‐simple sequence repeat markers and seedling life expectancy after Pc inoculation revealed significant different responses of C. sativa populations to Pc . The genetic variability observed within populations showed the potential response capacity of Spanish C. sativa populations to undergo fast adaptive evolution. The heritability value obtained for the “life expectancy” variable (h 2 = 0.21 ± 0.11) indicated that selection for resistance to Pc is possible. Genetic patterns reflected two evolutionarily meaningful groupings of populations, corresponding to the different selective pressure of the oomycete between sites. The differentiation coefficient obtained through markers classified as under neutral selection (F ST = 0.185) was lower than the quantitative genetic differentiation of “life expectancy” between C. sativa populations (Q ST = 0.682), providing evidence that selection acted spatially in a heterogeneous manner. A first link has been identified in trees between population structure and adaptive responses to pathogen‐induced selection. The study identified one marker under positive selection that could be used in marker assisted selection to predict resistance to Pc in non‐inoculated C. sativa trees.
To investigate the evolutionary relationships among the species of Trisetum and other members of subtribe Koeleriinae, a phylogeny based on DNA sequences from four gene regions (ITS, rpl32‐trnL spacer, rps16‐trnK spacer, and rps16 intron) is presented. The analyses, including type species of all genera in Koeleriinae (Acrospelion, Avellinia, Cinnagrostis, Gaudinia, Koeleria, Leptophyllochloa, Limnodea, Peyritschia, Rostraria, Sphenopholis, Trisetaria, Trisetopsis, Trisetum ), along with three outgroups, confirm previous indications of extensive polyphyly of Trisetum. We focus on the monophyletic Trisetum sect. Sibirica clade that we interpret here as a distinct genus, Sibirotrisetum gen. nov. We include a description of Sibirotrisetum with the following seven new combinations: Sibirotrisetum aeneum, S. bifidum, S. henryi, S. scitulum, S. sibiricum, S. sibiricum subsp. litorale , and S. turcicum ; and a single new combination in Acrospelion : A. distichophyllum . Trisetum s.s. is limited to one, two or three species, pending further study.












