Xiao‐Kai Fan, Jing Wu, Hans Peter Comes, Yu Feng, Ting Wang, Shu‐Zhen Yang, Takaya Iwasaki, Hong Zhu, Yun Jiang, Joongku Lee, and Pan Li
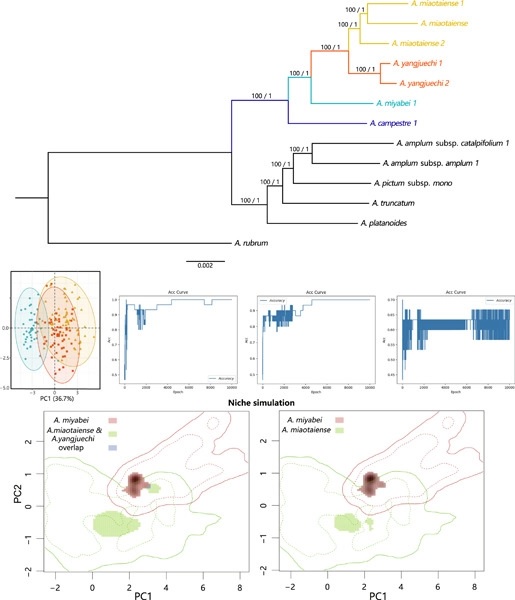
Accurate species delimitation is crucial for biodiversity conservation. The Acer series Campestria comprises four species, A. campestre L., A. miyabei Maxim., A. miaotaiense P. C. Tsoong, and A. yangjuechi Fang & P. L. Chiu. To clarify controversies over the taxonomic status of the latter three endangered species, we undertook phylogenomic, morphological, and niche differentiation analyses in series Campestria. Our coalescent species tree of 544 and 77 single-copy nuclear genes supported series Campestria as monophyletic, with A. yangjuechi having the closest relationship with A. miaotaiense. However, in the plastome-derived tree based on 64 protein coding sequences, the four species did not cluster together, and each of them grouped with some other sympatric Acer species. Given this nuclear-cytoplasmic conflict, we hypothesize that A. yangjuechi have been subject to nuclear gene introgression and plastid (pt) capture involving another sympatric maple, that is, A. amplum Rehder. Principal component analysis and machine learning based on morphological data could not separate A. yangjuechi and A. miaotaiense, but they both could be clearly distinguished from A. miyabei. Moreover, the niche overlap tests of the two more widespread species, A. miyabei and A. miaotaiense, showed they clearly occupy distinct niches. Overall, we conclude that A. miyabei and A. miaotaiense are distinct species, while A. yangjuechi (endemic to Mt. Tianmu/East China) should be treated as a subspecies of A. miaotaiense. Our study points out that multiple lines of phylogenomic, morphological, and ecological evidence prove highly useful in species delimitation. Additionally, our results should help to inform conservation measures for endangered species of the genus Acer/series Campestria in East Asia.