Antonio Giacò, Lucia Varaldo, Gabriele Casazza, Daniele De Luca, Paolo Caputo, Marco Sarigu, Gianluigi Bacchetta, Llorenç Sáez, and Lorenzo Peruzzi
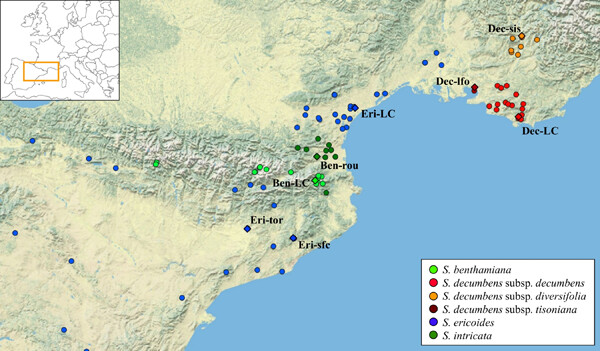
Santolina is a clear example of a genus lying in an alpha-taxonomic status, with species accepted only based on qualitative morphological descriptions. In particular, taxonomic issues still need to be resolved for Santolina populations from southern France and north-eastern Spain, and so, we carried out an integrative taxonomic study involving morphometrics, cypsela morphometrics, niche overlap, and phylogenetic analysis based on six plastid markers (trnH-psbA, trnL-trnF, trnQ-rps16, rps15-ycf1, psbM-trnD, and trnS-trnG). Our results revealed that the current taxonomic circumscription is not adequate. In particular, the Santolina populations at the foothills of eastern Pyrenees, previously included in the variability of Santolina benthamiana, have to be considered as a distinct species, namely, Santolina intricata. In addition, despite their high phylogenetic relatedness, S. benthamiana s.str. and Santolina ericoides can still be considered as distinct species due to clear morphological and ecological differentiation. Finally, we demonstrated that three different subspecies can be recognized in Santolina decumbens, a species endemic to Provence. For one of these subspecies, due to its extremely restricted distribution range, conservation issues are pointed out.
An integrative taxonomic approach involving morphometrics, niche analysis, and molecular systematics with plastid markers (trnH-psbA, trnL-trnF, trnQ-rps16, rps15-ycf1, psbM-trnD, and trnS-trnG) revealed new endemic taxa of Santolina in southern France and north-eastern Spain. In particular, Santolina intricata, previously considered as a synonym of Santolina benthamiana, has to be considered as a distinct species, endemic to eastern Pyrenees. Then, for Santolina decumbens, a species endemic to Provence, three subspecies can be recognized. Finally, despite a high phylogenetic relatedness, Santolina ericoides and S. benthamiana can still be considered as distinct species due to both morphological and ecological differentiation.