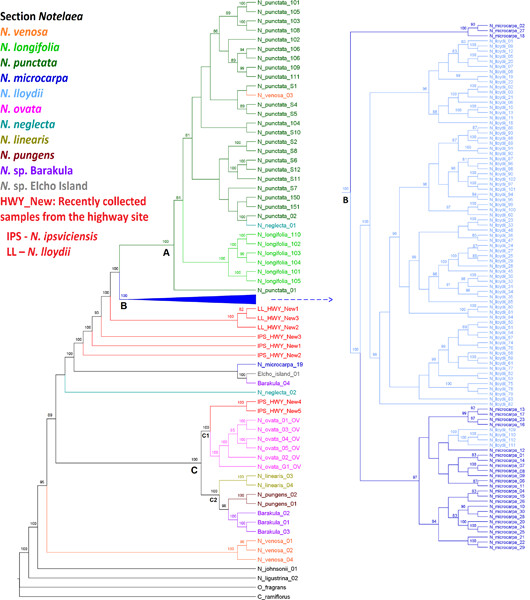
Ying‐Xue Jiao, Xiao‐Fan He, Rui Song, Xue‐Meng Wang, Han Zhang, Reziya Aili, Yue‐Hui Chao, Yu‐Hua Shen, Long‐Xi Yu, Tie‐Jun Zhang, and Shan‐Gang Jia
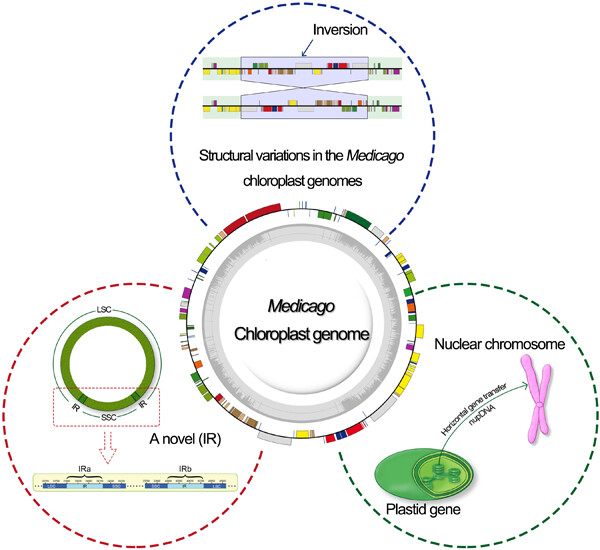
Medicago is a genus of legumes (Fabaceae) that resemble common clovers with pinnately trifoliate leaves and spirally coiled seed pods, and Medicago sativa is a famous forage crop throughout the world. In this study, we systematically assembled the complete plastid genomes of 18 Medicago species, representing 35 Medicago accessions, whose genome size ranged from ~119 to 125?kb, and identified one novel inverted repeat (IR) in two accessions of Medicago soleirolii (PI537242 and PI537243), albeit of no IRs in the most accessions. We built a phylogenetic tree based on common protein-coding sequences of 55 Medicago accessions in 38 species, which were placed into five clades with a divergence since 9.37 million years ago. Global alignment revealed independent genome evolution events, including eight inversions in nine species and four intron losses (ILs) in 10 species, among which four inversions and two ILs have not been reported previously. Within 109–111 unique genes, ndhA, rpl2, and ycf3 were under positive selection in 54 Medicago accessions. Finally, by aligning chloroplast genes against the nuclear genome assembly of M. sativa cultivar “Zhongmu No.1”, we found that a large number of chloroplast gene fragments were horizontally transferred to nuclear chromosomes in alfalfa, especially on the chr3:47518422–48722257 coordinates of chromosome 3. Our comprehensive exploration of Medicago chloroplast genomes provided insights for the understanding of Medicago diversity and their genomic evolution events.
1. Eight inversions were identified in 10 species, among which four inversions have not been reported previously.
2. A novel inverted repeat of ~0.3 kb was discovered in Medicago soleirolii in the present study.
3. Large numbers of chloroplast gene fragments were horizontally transferred to nuclear chromosomes in alfalfa.